We extend our gratitude to Dr. (Mrs.)
Anuradha Dubey, Scientist, Division of Parasitology, C.D.R.I.,

Drugs and Pharmaceuticals
Current R & D Highlights
(Leishmaniasis)
Contents
Features
·
Leishmaniasis: An
Overview 1
· Antimonial Therapy in Post- 7
kala-azar Dermal Leishmaniasis –
A Hobson’s Choice
· Novel and Validated Drug 11
Targets in Leishmania
· Approaches Towards Drug 22
Development for
Leishmniasis: A Review
· Mechanisms of Drug 35
Resistance in Kala-azar
· Is Vaccination Feasible 41
against Kala-Azar?
· Antileishmanial Potential 49
of Indian Medicinal Plants
Leishmaniasis: A Neglected 52
Tropical Disease
News & Views 60
R & D Highlights 62
R & D Technology 75
New Leads 78
Natural Products 83
Patents 95
CDRI Publications 101
We extend our gratitude to Dr. (Mrs.)
Anuradha Dubey, Scientist, Division of Parasitology, C.D.R.I.,
Leishmaniasis: An Overview
Mukesh Samant and Anuradha Dube
Division of Parasitology, Central
Drug Research Institute,
Leishmaniasis caused by protozoan parasites Leishmania, is a disease of poverty as its victims are among
the poorest. According to ranking after malaria it is a second most prevalent parasitic
disease. Leishmaniasis has been considered as a tropical affliction that
constitutes one of the six entities on the list of most important diseases of
World Health Organization/Tropical Disease Research (WHO/TDR) viz. Malaria,
Schistosomiasis, Filariasis, Chagas disease, African Trypanosomiasis,
Leishmaniasis, Leprosy, Tuberculosis.
1. History of Leishmaniasis:
Representations of skin lesions and facial deformities have
been found on pre-Inca pottery from
2. Risk Factors and
Definition of the Problem
In
The leishmaniases which causes considerable morbidity
and mortality is the collective name for a number of diseases which have
diverse clinical manifestations. Leishmaniases has traditionally been
classified in three major forms on the basis of clinical symptoms. The most
deadly form is visceral leishmaniasis (VL), which if left untreated, leads to
full-blown disease and invariably leads to death. A number of other species of Leishmania
cause cutaneous (CL) and mucocutaneous (MCL) leishmaniasis, which, if not
fatal, are still responsible for considerable morbidity of a vast number of
people in endemic foci. Leishmaniasis is spreading in several areas of the
world as a result of epidemiological changes which sharply increase the
overlapping of AIDS and VL.
This is the most common form of Leishmaniasis, also known as
‘Oriental sore’ which first appears as a persistent insect bite. Simple skin
lesions appear at the site of sand fly bite (fig 1) which self-heal within few
months but leaves scars. The incubation period can last from few days to
months. Gradually, the lesion enlarges, remaining red, but without noticeable
heat or pain. Resolution of the lesion involves immigration of leucocytes,
which isolate the infected area leading to necrosis of the infected tissues,
and formation of a healing granuloma in the floor of the lesion.
The disease is mostly prevalent in Mediterranean region,
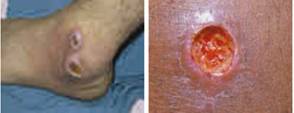
Fig1. Severe skin ulcer development
due to sand fly bite, a specific and primary symptom of cutaneous Leishmaniasis
followed by lesion enlargement, redness, but without noticeable heat or pain.
Variations of CL: Diffuse cutaneous leishmaniasis
(DCL)
This is a chronic, progressive, polyparasitic variant that develops
in context of leishmanial-specific anergy and is manifested by disseminated
non-ulcerative skin lesions, which can resemble lesions of lepromatous leprosy.
DCL is restricted to
Mucocutaneous Leishmaniasis (MCL)
This form of disease, also known as ‘‘espundia’’, causes
extensive destruction of naso-oral and pharyngeal cavities with hideous
disfiguring lesions, mutilation of the face and great suffering for life. MCL
is occasionally reported from
The causative agents of MCL in old world are L. aethiopica
(rare), and in new world are
L. braziliensis, L. guyanensis, L.
mexicana, L. amazonensis and L. panamensis.
VL is the most dreaded and devastating amongst the various
forms of leishmaniasis. VL is also known as Kala-Azar, Black Sickness, Black
Fever, Burdwan fever, Dumdum fever or Sarkari Bimari etc. It is the most severe
form of disease and if left untreated, is usually fatal. The parasite is
responsible for a spectrum of clinical syndromes, which can, in most extreme cases,
move from an asymptomatic infection to a fatal form of VL. It is characterized
by prolonged fever, splenomegaly, hepatomegaly, substantial weight loss,
progressive anemia, pancytopenia, and hypergammaglobulinemia (mainly IgG from
polyclonal B cell activation) and is complicated by secondary opportunistic
infections (Fig.2). The parasite invades and multiplies within macrophages
(free mononuclear phagocytic cells) and affects the reticuloendothelial system
including spleen, liver, bone marrow, and lymphoid tissue. The outcome of fully
developed VL is death, usually said to be due to concomitant infection
resulting from the weakened immunological state of the patient.
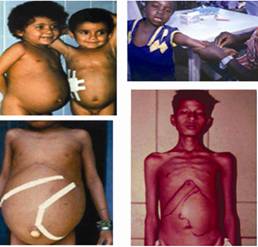
Fig.2.
Clinical
symptoms of visceral
leishmaniasis. Hepato-splenomegaly and substantial
weight loss
are main features.
VL is typically caused
by L. donovani complex, which includes three species: L. donovani
donovani, L d. infantum, and L. d. chagasi. L. donovani is
the causative in the Indian subcontinent and
There are more than 21 morphologically indistinguishable
species of Leishmania that infect humans. Conventionally, they are
classified and named mainly according to their geographical distribution and
clinical characteristics of the disease they afflict.
The Post Kala-azar Dermal Leishmaniasis (PKDL) is a
type of non ulcerative cutaneous lesion. After recovery from infection, VL
patients may develop a chronic form of CL i.e., PKDL which is developed in
about 10% of kala-azar patients generally one or two years after completion of
sodium stibogluconate (SSG) treatment and requires a long and expensive
treatment.
4. Geographical
Distribution of Leishmaniasis
Leishmaniasis occurs in 88 countries in tropical and temperate
regions, of which 72 are either developing or least developed. Approximately
1,98,000 people are affected with these diseases worldwide with 5,00,000
million new cases occurring each year, but the true picture remains largely
hidden since a substantial number of cases are never recorded. The
disability-adjusted life years (DALY) burden was 2,357,000 and total deaths
were 59,000 in 2001. It has been estimated that 90% of CL cases occur in 7
countries: Afghanistan, Algeria, Brazil, Iran, Peru, Saudi Arabia and Syria
whereas MCL
is endemic in Mexico and Central and South America (Fig 3).The annual estimate for the incidence and
prevalence of kala-azar cases worldwide is 0.5 million and 2.5 million,
respectively and of these, 90% cases occur in India, Nepal, Bangladesh and
Sudan. PKDL
is prevalent in
Global Status of Visceral Leishmaniasis
VL is endemic in 62 countries, with 200
million people at risk, an estimated 500,000 new cases each year worldwide and 41,000 recorded deaths
in the year 2000. The disease burden
associated with VL, measured in DALYs was estimated to be 1,980,000 (1,067,000
for male and 744,000 for female in year 2000. VL is caused by L. donovani in
the Indian subcontinent,
National Status Visceral Leishmaniasis
KA is present in
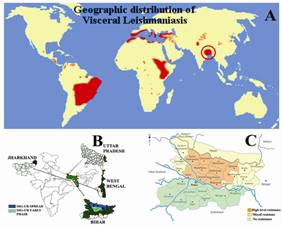
Fig.3. The worldwide distribution of visceral leishmaniasis (A),
VL affected states of India (B) and VL affected districts of Bihar (C).
5. Vectors of the Disease
Leishmaniasis is transmitted by the Phlebotomus spp. in the
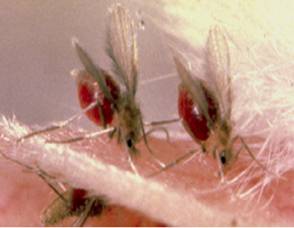
Fig.4. Sand fly, the vector host of Leishmania parasite
6. Morphology
and Digenetic Life cycle of Leishmania donovani
In
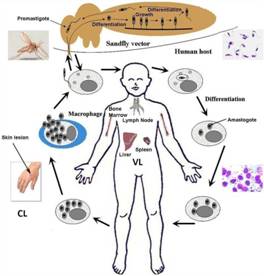
Fig.5. The life cycle of Leishmania.
Parasite shuttles between vector host, sand fly and human host. [Adapted from
Kumari et al, (2008)]
7. Factors Responsible for
Virulence and Survival of Parasite:
Cell surface glycoconjugates play a
pivotal role in parasite virulence and infectivity. Expression of complex and
unique glycoconjugates at the parasite cell surface appears to be crucial for
their survival and development in the sand fly vector and the mammalian host
macrophage. Sialoglycans as well as lipid-bound (LPG) and protein-bound (sAPs)
and (PPGs) phosphoglycan-containing glycoconjugates are the predominant
molecules on the cell surface and in the secretory products of the parasites
and are the target of intense research efforts.
8. Leishmania/HIV
Co-infections
Epidemiology of VL is further changing due to widespread
migration of population and emerging HIV/VL co-infection which is emerging as
an extremely serious problem. The risk of VL among AIDS patient’s increases by
100 to 1000 times in endemic areas as well as VL accelerates the onset of AIDS
in HIV infected people. To date, it has been reported
from 31 countries, with most of the cases from
9. Control
Strategies of the Disease:
Efficient case management based on early
diagnosis and treatment is the key to limit morbidity and prevent mortality.
Effective treatment of patients is also a measure to control reservoir and
transmission in anthroponotic foci, particularly for cases of PKDL, which are
thought to act as a long term reservoir of the disease. In addition, vector
control should be implemented wherever feasible. Spraying of houses with
residual insecticides has been an important measure in the past in
Based
on
1. Handman,
E., 2001. Leishmaniasis: current status of vaccine development. Clin Microbiol
Rev 14, 229-243.
2.
Palatnik-de-Sousa,
C.B., 2008. Vaccines for leishmaniasis in the fore coming 25 years. Vaccine 26,
1709-1724.
Views expressed in the journal are those of the
authors and the Editorial Board/Publisher takes no responsibility for the same.
We are a secondary abstracting service and the veracity of information is of
the source quoted and not our primary responsibility.
Editor
Antimonial Therapy in Post-kala-azar Dermal Leishmaniasis –A Hobson’s Choice
V.
Ramesh, Dhiraj Kumar and Poonam Salotra*
Department of Dermatology & Regional STD
Centre, and Institute of Pathology (ICMR), Safdarjang Hospital Campus, New
Delhi 110 029, India
Post-kala-azar dermal leishmaniasis (PKDL), an aftermath of
kala-azar (KA) or visceral leishmaniasis (VL) is an unusual dermatosis with
gross cutaneous lesions mainly comprising of hypopigmented macules, erythema,
and nodules. The lesions persist for long periods, and complications arise when
mucous membranes are affected, the most serious being blindness due to ocular
involvement. The disease is relatively common in the Indian subcontinent (
The incidence of PKDL has important implications in
transmission of VL, as PKDL provides the only known reservoir of the parasite
in
So far, little is known about the factors of parasite/host
origin that drive the parasite to cause a shift in the site of infection from
viscera to dermis and thereby the clinical manifestation of the disease. It is
not known whether the parasite in PKDL lesions is the residual parasite after
VL infection or is introduced upon re-infection by sand fly vector. Reactivation of the persistent infection is
considered to underlie PKDL pathology. Genes showing up regulation in PKDL like
major surface proteins gp63, PSA-2 and Amastin, may be the factors that
contribute to persistence after VL and play a role in altered clinical
manifestation in PKDL. A role of cytokines IFN-g,
TNF-a and IL-6 is implicated in
distinct PKDL pathogenesis. Interference with type 1 effector activity in PKDL
may be due to minimal expression of IFN-gReceptor1
or simultaneous presences of elevated levels of IL-10, IL-6 and TGF-b
that have counter acting activities.
Upon treatment, the restoration of IFN-gR1,
coupled with down regulation of counter acting cytokines, facilitates the
action of signals associated with IFN-g,
yielding parasite clearance. Higher expression of chemokines (MCP-1, MIP-1a,
MIP-1b) in PKDL indicates a role
of these molecules in parasite perseverance by host cell recruitment at the
site of infection. Distinct Immune profile is observed in Sudanese PKDL patients
where IL-10, IFNg and IL-4 present in
majority of tissue lesions, while in Indian PKDL here is preponderance of IFNg,
TNFa, IL-10, TGFb,
IL-6 and IL-4 in lesion tissues. In Sudan VL cases with high level of IL-10 are
prone to develop PKDL, while in
Intriguingly, treatment of PKDL requires sodium antimony
gluconate (SAG) therapy for a duration exceeding 4 times that required for
treatment of KA, at the same dose. The chequered role of antimony in medicine
has reached a climax in the leishmaniases, lending credence to the pithy remark
made almost 2 decades ago that apart from antimonials no other heavy metal
treatment in any disorder has enjoyed such a reputation and remained unchanged
over decades. KA is the severest form in which pentavalent antimonials
commendably scaled down the mortality to as low as 5% from that of 95% in the
pre-antimony era. Being poorly absorbed
orally, the mode of administration of antimony is parenteral. In contrast to
the trivalent form, pentavalent compounds are less toxic, high doses can be
given as they are quickly excreted, and the sodium salt is better tolerated
than the potassium one. Acquired resistance and relapse led experts to
recommend 20mg/kg/day to a maximum of 850mg , 8.5 ml (10mg/ml) of sodium
stibogluconate (SSG) or 10 ml (85 mg/ml) of meglumine antimoniate, in KA for a
minimum of 20 days, which could extend
up to 40 days if required. Common side-effects were stated to be anorexia,
vomiting, nausea, malaise, myalgia, headache and lethargy, followed uncommonly
by electrocardiographic changes, and rarely renal damage. A subsequent review
into toxicity and efficacy of SSG in various studies recommended that the dose
of 20mg/kg/day for 28 days could be routinely used without an upper limit, with
a provision to extend if required.
Summing up the use of SSG during latter half of previous
century in highly endemic areas of
India, it was seen that efficacy had diminished over years as treatment
in some had to be prolonged to achieve
cure, while some did not respond indicating that resistance had developed
probably due to exposure to suboptimal doses in the past. More importantly, ECG
changes were seen in a significant number of patients, some of whom died of
cardiac causes. Reviewing current situation and regional variations in the
responsiveness of KA in
This regimen of SSG for 4 months administered intramuscularly
is tolerable in PKDL, with mild ECG changes and raised plasma urate with
arthralgia ocassionaly, none of which precluded completion of therapy. In some
cases the duration needs to be extended
to 200 days to combat refractoriness and relapse, and also to ensure subsidence
in those with extensive lesions. ECG
abnormalities revert to normal on
stopping the drug, and arthralgia, pain
and swelling at site of injection are managed with analgesics and brief
discontinuation of therapy; neuritic symptoms and change in taste were
occasionally encountered. Between 1997
till 2007 a total of 126 cases of PKDL, 112 adults and 14 children have been
treated at our centre in
When treatment rallies around a month like in KA it is
tolerable, but if prolonged, rheumatic side-effects like myalgia and joint
pains dominate, which make irregular therapy inevitable. Studies have
surprisingly commented little on the relative lack of cardiac side-effects in
PKDL, despite the long duration of therapy with SSG as compared to KA. It may
relate to systemic disease and general condition of the patient in KA.
Pancreatitis, mainly subclinical and unusually rare in those with KA or PKDL,
has occurred during SSG therapy for cutaneous leishmaniasis as evidenced by
increased serum amylase or lipase levels associated with signs or symptoms of
abdominal discomfort but patients could complete treatment. A recent study
inferred that both hyperamylasemia and raised liver transaminases were
clinically insignificant and not deterrents to treatment. Fatalities due to
pancreatitis have occurred in those co-infected with HIV. Apart from drug
resistance, the varying clinical response and toxicity of SSG may be related to
a bimodal type of drug clearance in the patient accounting for rapid and slow
eliminators. Higher amount of antimony
than that specified by the manufacturers or traces of the more toxic trivalent
compound in the preparation could also account for toxicity.
Reducing the volume injected by increasing concentration of
antimony could minimize local side-effects. Unfortunately at levels above
100mg/ml SSG tends to supersaturate forming crystals and precipitates.
Alternatively, duration of therapy could be shortened by immunotherapy in
addition to the recommended dose of SSG and more studies are required in this
direction. Liposome entrapped Pentostam achieved better results with low doses
in experimentally infected mice with L
donovani indicating enhanced drug delivery, but this was not further
perfected and popularized for wider use. Pending a good multi-drug formulation,
an acceptable therapy for PKDL is required. Since the duration is likely to be
longer than in KA and the patients are in otherwise good health without the
need for hospitalization, oral therapy is preferable to ensure compliance.
Currently miltefosine, the only drug to fulfill this requirement, is beset with
high cost of therapy and is not freely available as yet. Cure of antimony
unresponsive Indian PKDL has been
obtained with oral miltefosine at a dose of 50mg twice daily for 3 months or at
50mg thrice daily for 2 months. l
miltefosine treatment is also successful in PKDL with HIV co-infection. However
results of Miltefosine trials in PKDL are awaited to find out the optimum dose
and duration. Amphotericin B, both in the liposomal and non-liposomal forms has
been well tried in KA, but in PKDL more studies are required to recommend the
adequate dose and duration of therapy. Till then antimonial appears to be the
Hobson’s choice with amphotericin B in reserve for refractory or
antimony-resistant patients of PKDL.
Novel and Validated
Drug Targets in Leishmania
Uma Roy, Kishore Kumar and Prachi Bhargava
Department
of Biochemistry, Central Drug
Research Institute,
1.
Introduction
Leishmania are protozoan parasite(s) that causes a wide spectrum
of clinical manisfestation in human(s) collectively referred to as
leishmaniasis ranging from self healing cutaneous (CL), mucocutaneous (MCL)
skin ulcers to life threatening visceral diseases. (VL or kala azar)(Murray et al., 20051). Leishmaniasis
is a devastating disease that affects about two million people each year and
threatens one fifth of the world’s population, new treatments are desperately
needed (Murray et al., 2005).
The Leishmania parasites are transmitted by an
invertebrate sandfly vector, Phlebotomus and exist in two major
developmental stages. Infected sandflies introduces the metacyclic forms of the
promastigote stage into the bloodstream of the vertebrate host. The
promastigote differentiates into amastigote stage that is adapted for survival
in the phagolysosome of the macrophages, the cell responsible for pathogen
elimination. During promastigote to amastigote differentiation, the parasites
are subjected to drastic environmental changes, including sharp rise in
temperature, a drop in extracellular pH, an increased exposure to oxygen and nitrogen
reactive species, an extracellular proteolytic activity and nutritional
starvation (Barak et al.,2005). The association of
Leishmaniasis with HIV has also been reported from 33 countries, where up to
70% of potentially fatal visceral leishmaniasis (VL) cases are
associated
with HIV infection, and up to 9% of AIDS cases suffer from newly acquired or
reactivated VL. No effective vaccines are available against Leishmania infections
as yet (Handman, 2001) and treatment relies solely on chemotherapy with
pentavalent antimonials as first-line drugs and amphotericin B and pentamidine
as second-line drug (
The route to drug target identification
has been through comparative biochemistry of host and parasite enzymes,
metabolites or protein identified in parasite. Biochemical analysis, genome
sequencing of three Leishmania species (L. major, L. braziliensis, L. infantum) have identified potentially
useful target enzymes, transporters, metabolites, hypothetical proteins that
are distinct to parasite and their mammalian host.
2. Comparision of three Leishmania
genome
Genome sequences of three species of Leishmania (L. major, L.
infantum and L. braziliensis) are now available at
gene database (http://www.genedb.org). The chromosomes of Leishmania differ from those of the trypanosome species in not
having extended telomeric regions containing species specific genes (Peacock et al., 2007). The complete genome of L.
major, L. infantum and L. braziliensis has provided many new
potential targets to be used in conjunction with comparison and functional
genomic studies. Such comparative genomic study will allow us to identify the
molecules or biochemical pathways that have been successfully targeted in other
pathogens. The genome mining will also aid in large scale proteomics studies
generating expression profiles of Leishmania parasites and gene targets
for treatment development.
Comparison of L. braziliensis and L. infantum revealed marked conservation of synteny and
identified only ~200 genes with a differential distribution between the three
species. L. braziliensis, contrary to Leishmania
species possesses components of putative RNA mediated interference (RNAi)
pathway, telomere associated transposable elements and spliced leader
associated (SLACS) retrotransposons. Differentially distributed genes between
the species encodes protein for parasite survival in the macrophages and
pertaining to host parasite interaction (Peacock et al., 2007). The genome sequence have given us a shortcut to a
small number of largely novel genes
‘‘Given their lack of similarity to human genes, they present a limited
repertoire of potential targets for drugs and vaccine development allowing
researchers to optimize the use of limited resources”. Cyclopropane fatty acid
synthase (CFAS) an enzyme that may be involved in producing components of the
cell membrane, is present in the genome of L. braziliensis and L. infantum, but is absent in the human
genome. CFAS is also involved in virulence and persistence in Mycobacterium,
causative agent of tuberculosis, so the identification of this potential target
CFAS gene in Leishmania raises the exciting possibility that some
virulence factor are conserved between bacterial and eukaryotic intracellular
pathogens (Peacock et al., 2007).
Biological studies for the function of 50% of Leishmania genes are
lacking. The comparitive genome study would provide a route to find those that
might be essential to each species (Rochette
et al., 2008)
3. Thiol
Metabolism
The enzymes of thiol metabolism and in some cases the thiols
themselves, of parasitic protozoa differ in many interesting ways from those of
mammals. Trypanosoma and Leishmania are most
remarkable in that they have trypanothione reductase (TR) instead of
glutathione reductase (GR).
This enzyme is responsible for maintaining the parasites, reducing
intracellular milieu by keeping trypanothione [N1, N8-bis-(glutathionyl)
spermidine] in the dithiol state. The crucial role of TR for thiol homeostasis
and its absence from mammalian cells suggest that it might be well suited as a
target molecule for rational drug development. The
trypanothione system, which replaces the nearly ubiquitous
glutathione/glutathione reductase (GR) system, protects the parasites from
oxidative damage and toxic heavy metals, and delivers the reducing equivalents
for DNA synthesis. The parasite system is far
less efficient than mammalian glutathione peroxidases in detoxifying
hydroperoxide, but has the advantage of much broader substrate specificity,
with lipid hydroperoxides also being reduced. The relatively low activity of
the tryparedoxin peroxidase system is in
accordance with the high sensitivity of the parasites to oxidative stress.
Trypanosomes and Leishmania have superoxide dismutase (SOD), but lack
catalase and glutathione peroxidase. Thus, the trypanothione system seems to be
the only mechanism to detoxify hydrogen peroxides.
3a.Trypanothione
Reductase
Trypanothione is kept reduced by the flavoenzyme TR. Several
reverse genetic approaches have undoubtedly shown that TR is essential in
different Leishmania species as well as in bloodstream of T. brucei
(Krieger, 2000) and is thus
an attractive target molecule for structure-based drug design. Within the past
15 years, numerous compounds have been elucidated that inhibit TR, but not
human GR, which is the closest related host enzyme. Despite knowledge of the
three-dimensional structure of the protein and of complexes with its substrates
and an inhibitor, as well as several high-throughput and virtual screening
approaches, inhibitors of TR that are suitable to enter the clinical phase are
still elusive. This lack of success might be attributable to several factors.
The extremely wide active site of the parasite enzyme represents an obstacle
for a structure-based drug design. In addition, the pharmacokinetic properties
of the potential inhibitors are crucial because of insufficient uptake, rapid
extrusion or metabolism play significant roles in determining the in vivo
efficacy of a drug. Another important point is the in vivo half-life of
a target enzyme, and this has not been determined for TR in any of the trypanosomatid
species. Thus, it is still not clear if reversible, irreversible or turncoat
inhibitors are likely to be the most promising candidates.
3b.Thioredoxin
reductase
Thioredoxin reductase (TrxR) is a pyridine
nucleotide-disulphide oxidoreductase as are GR, TR, and lipoamide
dehydrogenase. TrxR maintains the levels of reduced thioredoxin, a protein
involved in the activity of ribonucleotide reductase, transcription factors and
cell signaling, and the detoxification of reactive oxygen species. Most studies
to date on TrxR of parasitic protozoa have concerned the enzyme of P.
falciparum. Current evidence suggests that it is a promising drug target,
although validation is awaited. Interestingly, P. falciparum TrxR
differs from the human enzyme in not only containing selenocysteine, but having
a C-terminal cysteine pair separated by four amino acids. The unusual nature of
the C-terminal domain prompts thoughts on whether it has a distinct role in the
parasite e.g., as a thiol that acts as a general reductive reagent in the cell
and so a special adaptation of the parasite for counteracting oxidative stress)
and how it interacts with the parasite’s thioredoxin (which is presumed, but
yet to be discovered (Krauth-Siegel and Coombs, 1999).
4. Folate Metabolism
A biochemical pathway that has been exploited in the past for
the treatment of infectious disease is the folic acid biosynthetic pathway.
Inhibitors of folate metabolism are known to be important for malaria, bacteria
and cancer chemotherapy. Perturbation in the folate pathway analog which
inhibits the mTHF recycling pathway rapidly leads to nucleotide imbalance and
thus causing cell death. Leishmania cannot synthesize folate de novo
(e.g. - folate and pterin) and must import these metabolites from an exogenous
source (Ouellette et al., 2002). A
novel class of transport membrane proteins is responsible for their uptake
(Richard et al., 2002; Richard et al., 2004; Cunningham et al 2001).
Enzymes of the total Leishmania folate pathway have been studied and
these include the bifunctional dihydrofolate reductase-thymidylate synthase
(DHFR-TS) and the folyl polyglutamate synthetase (El Fadili et al., 2002; El fadili et al., 2003). The genome sequencing of Leishmania
species (Ivens et al., 2005), http:/www.genedb.org
and their analysis have highlighted the presence of several proteins implicated
in folate metabolism (Ouellette et al.,
2002). L.major genome sequence showed the presence of two isoforms of SHMT
gene cytosolic and mitochondrial suggesting the folate metabolism of Leishmania
to be compartmentalized (Gagnon et al.,
2006). For better understanding of the properties of SHMT, and complexities of
folate metabolic pathway of Leishmania the enzyme has been overexpressed
and characterized in L. donovani (Vatsyayan and Roy, 2007). The folate pathway provided a valuable
target for microorganisms such as bacteria and plasmodium for drug
intervention. However till date no drug targetting the folate pathway have been
found to be effective against Leishmania infection. There seems to be
two reasons for this, first Leishmania cannot synthesize folate
(biopterin and folate) de novo and must import these metabolites from exogenous
source (Ouellette et al., 2002).
Secondly the enzymatic reduction of folate to become active as tetrahydrofolate
(THF), a coenzyme required for one carbon (C1)transfer reactions, can be
catalyzed by both DHFR-TS and pteridine reductase1(PTR1). When DHFR-TS is
inhibited PTR1 is overexpressed, hence it is necessary to block both DHFR-TS
and PTR1 for effective interference with folate metabolism (Opperdoes and
Coombs, 2007). Several genes that encode enzymes of folate biosynthesis are not
in the Leishmania genome but twelve genes that encode a novel class of
membrane transport protein responsible for folate transport has been reported.
Putative SHMT inhibitors, including thiosemi-carbazide,
have poor activity against Leishmania but further work may lead to more
potent inhibitors. The development of antifolate drugs for leishmaniasis treatment
requires further studies of key enzymes of this pathway
5. Glycolytic Pathway
In all kinetoplastida studied so far the majority of the
glycolytic enzymes are localized in organelles called glycosomes, whereas in
other organisms these are cytosolic. In blood stream form of T. brucei
glycolysis is the main source of energy as they lack functional Krebs cycle
while in Leishmania and T. cruzi the glycolytic pathway is less
important but because their glycosome contain important anabolic and
anapleuratic pathways that are interdependently linked to glycolysis, as any
compound designed to act against African trypanosomes may also be effective
against these parasites. Due to this compartmentation, many regulatory
mechanisms operating in other cell types cannot work in trypanosomes. This is
reflected by the insensitivity of the glycosomal hexokinase (HK) and
phosphofructokinase (PFK) to compounds that act as activity regulators in other
cell types (Bakker et al., 2000;
Michels et al., 2000). Blocking of
Parasite enzyme without producing damage to glycolysis in host remains
challenging. Several approaches can be considered- 1) Exploitation of metabolic
differences 2) Exploitation of differences in 3 D structure 3) Exploitation of
unique reactive residue in or near the active site of the parasite enzyme
5a.Hexokinase
Hexokinase (HK) catalyzes the conversion of glucose to glucose
6-phosphate. The sequence of hexokinase from L. major was found to encode an enzyme with a molecular mass of
51.74 kDa. The L. major genome was found to have two copies of
hexokinase coding sequences in tandem with an intergenic spacer of 2.58 kb. The
HK gene was transcribed in large amounts in the promastigote stage, whereas
there is only weak expression in the amastigote stage as determined by RT-PCR
analysis (Umashanker et al., 2005). HK was also
purified from L. mexicana from glycosome of promastigotes. The specific
activity increased with the ageing of promastigote culture (Pabon et al., 2007)
5b.Glucose 6
phosphate isomerase
Glucose-6-phosphate isomerase (PGI) is an intracellular enzyme
that catalyzes the reversible conversion of D-glucose
6-phosphate (G6P) to D-fructose 6-phosphate (F6P). The
native Leishmania PGI is a homodimeric molecule of
60 kDa per monomer with 47% sequence identity to human PGI (Cordeiro et al., 2004).
5c.Phosphofructokinase
Phosphofructokinase (PFK) catalyzes the phosphorylation of
fructose 6-phosphate (F6P) to fructose 1, 6-bisphosphate (FBP) in an
essentially irreversible reaction. The gene of PFK has been cloned and
characterized from L. donovani and T. brucei. L. donovani has a single PFK gene
copy per haploid genome that encodes a polypeptide with a deduced molecular
mass of 53.988 kDa while human enzyme has subunit of 85 kDa .The predicted
amino acid sequence contains a C-terminal tripeptide that confirms to an
established signal for glycosome targeting. L. donovani PFK showed most
sequence similarity to inorganic pyrophosphate (PPi)-dependent PFKs,
despite being ATP-dependent. It thereby resembles PFKs from other
Kinetoplastida such as T.brucei, T.borreli (Lopez et al., 2002). Furanose sugar amino amides as a novel class of
inhibitors for both enzymes with IC50 values of 23 μM
against T. brucei PFK. The residue
5d.Triose
Phosphate Isomerase
Triose Phosphate Isomerase (TIM) is an important enzyme of
glycolytic pathway which interconverts glyceraldehyde 3- phosphate to
dihydroxyacetone phosphate. The TIM gene from L. donovani was cloned,
over expressed, analyzed and submitted to data bank (Kumar and Roy, 2006
Accession no- DQ649411). Homology search showed 88.10, 66.67, and 49.2 %
identity with L. mexicana, T.cruzi, and Human respectively. In L. donovani Glutamate was found at
position 66 instead of Glutamine. The dimer of TIM is quite stable and is the
active form of the protein (Knowles and Albery, 1977). In fact, there are several reports that
suggest that mutations at the subunit interface of the protein destabilize the
dimer leading to either complete inactivation or drastic decrease in the
activity of the enzyme. The knock out of this enzyme in T. brucei established that TIM is also a vital enzyme as it would
lead to complete suppression of growth arrest (Sandra Helfert and Christine
Clayton,
5e.Glyceraldehyde
3- phosphate Dehydrogenase
Glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) catalyzes the conversion of glyceraldehyde 3-phosphate to
D-glycerate 1, 3-bisphosphate. This
enzyme is homotetrameric and it appears that the active site of the enzyme and
the neighboring nicotinamide binding site for NAD+ are well
conserved (Verlinde et al., 2001).
However, the binding site for the adenosine portion of NAD+ exhibits
significant differences between parasite enzyme and host. This difference can
be exploited for designing selective drugs.
6. Polyamine Pathway
The polyamine pathway of protozoan parasites has been
successfully targeted in anti parasitic therapies. Polyamines
are ubiquitous organic cations found in virtually every eukaryotic cell and
plays critical role in key cellular processes such as growth, differentiation
and macromolecular biosynthesis. All the gene of polyamine pathway i.e
ornithine decarboxylase (ODC), S-adenosylmethionione decarboxylases (AdoMetDC) and
spermidine synthase (SPDS) has been cloned and their knockouts by gene
replacement has demonstrated the essential role of each of these enzymes in L. donovani (Roberts et al., 2001; Roberts et al., 2002) unless they can access
sufficient amount of exogenous putrescine and spermidine but the parasites
exhibit negligible uptake capacity (Hanson at al., 2005). However polyamine
transport itself has been characterized at biochemical level in various
protozoa (Basselin, 2000). The inhibition of any of the polyamine the parasite
cannot synthesize trypanothione, a conjugate of spermidine and glutathione that
is unique to Trypanosoma and Leishmania.
Trypanothione is a reducing agent with many protective and regulatory functions
and consequently its depletion is detrimental to the parasites. Recent studies
on polyamine supplementation shows that L.
donovani lacks an intact back conversion pathway thus the pathways
operating in promastigotes stage of parasite differ crucially from that in the
host.
Enzymes involved in
polyamine pathway of Leishmania are:
6a.Arginase
Ornithine, the first amino acid from which polyamines are
generated is produced from arginine by arginase enzyme. An arginase activity
has been detected in L. mexicana and L. amazonansis, while T. cruzi and T. brucei lacks arginase
activity. Arginase provides a building block for
production of polyamines so it has been touted as a potential antileishmanial
drug target, because N(omega)-hydroxyarginine, an inhibitor of arginase that is
produced by the macrophages during the formation of nitric oxide, can reduce
polyamine levels in Leishmania amastigotes and lowers parasitic loads (Iniesta
et al., 2001).
The lethal nature of arginase knockouts establishes that Leishmania promastigotes have only a single avenue for ornithine
biosynthesis (Roberts et al., 2004). The arginase gene from L. donovani has
been cloned, analyzed and submitted to data bank (Roy et al., 2006 Accession no-DQ649412). Complete ORF codes for 330
amino acids with GC content of 60.3%. Homology search of L. donovani
with L. mexicana and Human showed
99%, and 42 % identity
respectively.
6b.Ornithine
decarboxylases
Ornithine decarboxylase is the first
enzyme of polyamine pathway, catalyzes ornithine to putrescine. It is also
validated as drug target because of major differences between parasite ODC and Human ODC. The parasite ODC is quite
stable (half life > 6 hr in T. brucei and > 20 hr in L.
donovani) as compared to human ODC ( half life < 1 hr). ODC knockouts
were incapable of growth in polyamine deficient medium. The DFMO and
3-aminoxy-1-aminopropane (APA) are the strong inhibitors of ODC. In recent
studies it was observed that L. donovani
ODC overexpression exhibited significant resistant to sodium stibogluconate (Singh et al.,
2007).
6c.S-adenosylmethionione
decarboxylases
S-adenosylmethionione decarboxylases (AdoMetDC) generate
decarboxylated S-adenosylmethionione (dcAdoMet) which serves as the aminopropyl
group donor for spermidine and spermine. The latter is absent from Leishmania. A very potent and selective
irreversible inhibitor of AdoMetDC is 5
6d.Spermidine
synthase
Spermidine biosynthesis is catalyzed by spermidine synthase
which transfers an aminopropyl moiety from decarboxylated S-adenosylmethionione
to putrescine. The L. donovani spermidine
synthase is present as a single copy gene in genome exhibiting 56% identity
with human. Two sequences encoding the spermidine metabolizing enzymes
deoxyhypusine synthase and homospermidine synthase have been isolated from L. major and published in gene bank. The product of these two genes
are excellent drug targets (Akopynts et
al., 2001). S-adenosyl-1, 8-diamino-3-thiooctane (AdoDATO) is potent
inhibitor of T. brucei spermidine
synthase.
7. Sterol Biosynthetic
Pathway
Isoprenoid compounds are ubiquitous in prokaryotic and
eukaryotic cells with the sterols usually the most abundant isoprenoid group
present in eukaryotes. Sterols perform a structural function as constituents of
cellular membranes and this has been referred to as their ‘bulk membrane’ role. The most extensively examined parasitic protozoa as far as
sterols are concerned are the trypanosomatids. It has emerged that these
parasites have close similarities to fungi in relation to their sterol
composition and sterol biosynthesis. This has offered an opportunity for the
development of chemotherapy by targeting the sterol biosynthetic pathway using
the types of drugs already successfully employed against fungal pathogens.
The enzymes of this
pathway are attractive targets for the specific treatment of leishmaniasis,
because the etiological agents for the disease that is the leishmanial
parasites have a strict requirement for specific endogenous sterols (ergosterol
& analogs) for survival and growth and cannot use the abundant supply of
cholesterol present in their mammalian host. There are differences in the
enzymes in the biosynthetic pathways of ergo sterol and cholesterol. A number
of enzymes in the ergosterol biosynthetic pathway have been investigated as
potential drug targets for these organisms and have shown great promise. Thus,
C14a-demethylase, sterol
24- methyltransferase, 3-hydroxy-3-methylglutaryl CoA reductase,
squalene epoxide, squalene synthase and farnesyl pyrophosphate synthase have
been studied both individually and in combination, with varying degrees of
success (Lorente et al., 2005).
Ergosterol biosynthesis inhibitors with potent in vitro activity and special pharmacokinetic properties in mammals
can induce radical parasitological cure in animal models of several forms of
leishmaniasis (Urbina, 2002).
7a.S-adenosyl-L-methionine:
Delta (24 (25))-sterol methenyltransferase
Trypanosomatids contain predominantly ergostane-based sterols,
which differ from cholesterol, the main sterol in mammalian cells, in the
presence of a methyl group in the 24 position. The methylation is initiated by
S-adenosyl-L-methionine: Delta (24 (25))-sterol methenyltransferase, an enzyme
present in protozoa, but absent in mammals. The importance of this enzyme is
underscored by its potential as a drug target in the treatment of the
leishmaniasis (Carmen Jiménez-Jiménez, 2008).
The C-24 transmethylation reactions involving S-adenosyl methionine as
the methyl donor and a Δ24(25)-sterol or Δ24(24′)-sterol
substrate can be inhibited by various azasterols with a nitrogen substitution
in the side chain and such compounds have been tested against trypanosomatids
(Roberts, 2003).
7b.Sterol C14
alpha-demethylase
Recent work with the sterol C14 alpha-demethylase inhibitor
D0870, a bis triazole derivative, showed that this compound is capable of
inducing radical parasitological cure in murine models of both acute and
chronic Chagas' disease. Other inhibitors of this type, such as SCH 56592, have
also shown curative, rather than suppressive, activity against T. cruzi in
these models. Leishmania species have
different susceptibilities to sterol biosynthesis inhibitors, both in vitro and in vivo. L. braziliensis
promastigotes, naturally resistant to C14 alpha-demethylase inhibitors such as
ketoconazole and D0870, were susceptible to these drugs when used in
combination with the squalene epoxidase inhibitor terbinafine (Urbina, 1997).
7c.3-hydroxy-3-methylglutaryl
CoA Reductase
In eukaryotes the enzyme 3-hydroxy-3-methylglutaryl CoA
(HMG-CoA) reductase catalyses the synthesis of mevalonic acid, a common
precursor to all isoprenoid compounds. This protein from Leishmania lacks the membrane domain characteristic of eukaryotic
cells but exhibits sequence similarity with eukaryotic reductases. In Leishmania HMG-CoA
reductase is up-regulated when sterol synthesis is inhibited by drug pressure
and this activation is apparently performed via post-transcriptional control (Montalvetti
et al., 2000). The lack of
sensitivity to mevalonate and sterols is consistent with the absence of a
membrane domain and may be a consequence of unique biological properties of the
isoprenoid biosynthetic pathway in protozoa. Trypanosomatids are early
branching eukaryotic cells and their cell organization differs considerably
from that of mammalian cells ( Peña-Díaz et
al., 1997) Specific features present in trypanosomatids but absent from
their hosts may be exploitable in providing targets for rational drug design .
7d.Squalene
Synthase
Squalene synthase catalyzes the first committed step in sterol
biosynthesis and is currently under intense study as a possible target for
cholesterol-lowering agents in humans, but it has not been investigated as a
target for anti-parasitic chemotherapy. Growth inhibition and cell lyses
induced by Hydroxy biphenylquinuclidines (BPQ-OH) an inhibitor in both
parasites (L.mexicana and T.cruzi) was associated with complete depletion of
endogenous squalene and sterols, consistent with a blockade of de novo sterol synthesis at the level of
squalene synthase. Ultra structural analysis of the treated parasites revealed
several changes in the morphology of promastigote forms. The main ultra
structural change was found in the plasma membrane, which showed signs of
disorganization, with the concomitant formation of elaborated structures.
Alterations in the mitochondrion-kinetoplast complex such as mitochondrial
swelling, rupture of its internal membrane and an abnormal compaction of the
kinetoplast were also observed. Other alterations included the appearance of
multivesicular bodies, myelin-like figures, alterations of the flagellar
membrane and presence of parasites with two or more nuclei and kinetoplasts (
Rodrigues et al., 2005). The squalene
synthase gene from L. donovani was cloned, analyzed and submitted to
data bank (Bhargava and Roy, 2006 Accession no- AM229310). The 1245 bp
confirmed clone contained an open reading frame of 415 amino acids giving a
predicted mass of 47.35 KDa. Comparision of the LdSSN deduced amino acid
sequence with SQS from different species showed the highest identity with Leishmania major (91%), followed by T.cruzi (57%), T.bruzi (48%), Mus musculus
(45%) and human (44%). The two signature sequences of squalene synthases were
present at position 164-179 and at 200-227. The secondary structure prediction
showed that it consists of 40.10% of alpha helix and the GC content is 59%.
7e.Farnesyl
Pyrophosphate Synthase
The sensitivity of trypanosomatid protozoa to isoprenoid biosynthesis
inhibitors (Docampo et al., 2001)
offers a unique opportunity for drug target identification and the
subsequent development of new anti-trypanosomatid agents. Farnesyl
pyrophosphate synthase (FPPS) plays a central role in metabolism through the enzymatic
generation of FPP, which is used for protein prenylation, for the
synthesis of sterols, dolichol, heme a, and ubiquinone, and
is potently inhibited by bisphosphonates. Stringent genetic
validation of putative drug targets is desirable before the rational
design of inhibitory compounds intended for chemotherapeutic use is
undertaken. Studies validate FPPS as a drug target through the use
of RNAi. It provides genetic evidence that FPPS plays an essential
cellular role in T. brucei and demonstrates that the
enzyme is vital for parasite survival in vitro and in vivo.
The finding that a similar pharmacophore can be obtained by
structure-activity investigations of in vitro growth and enzyme inhibition
data further validates T.brucei FPPS as the target of bisphosphonates ( Montalvetti et al., 2003)
8. Glyoxalase
Pathway
The glyoxalase pathway is the main catabolic pathway of
methylglyoxal, a toxic 2- oxoaldehyde which occurs in all living cells as a by
product of glycolysis through reaction catalyzed by triose phosphate isomerase.
It first reacts non enzymatically with one or both thiol of trypanothione
forming hemiacetal. These hemiacetals are the substrates for glyoxylase pathway
forming D- lactate as final product (Silva et
al., 2005). In L. infantum it has
been shown that enhancement of methylglyoxal or depletion of trypanothione
leads to significant increase in the concentration of this toxic compound hence
this data might be useful for research drug targeting this disease (Lages et al., 2007)
9. Topoisomerase
Topoisomerases are enzymes that use DNA strand scission,
manipulation and rejoining activities to directly modulate DNA topology. These
actions provide a powerful means to effect changes in DNA super coiling levels
and allow some topoisomerases to both unknot and decatenate chromosomes. They
are involved in replication, transcription, chromosomal condensation and
segregation and many other vital cellular processes.
DNA topoisomerases are the primary targets of many antitumour drugs. DNA topoisomearases
are the key enzymes involved in carrying out high precision DNA transactions
inside the cells. However, they are detrimental to the cell when a wide variety
of topoisomerase-targeted drugs generate cytotoxic lesions by trapping the
enzymes in covalent complexes on the DNA (Majumdar et al., 2006). Many
antiparasitic compounds have been found to act via topoisomerases having more
profound effect on the parasite protein than the host. The identification of
DNA topoisomerases as a promising drug target is based on the clinical success
of camptothecin derivatives as anticancer agents. The recent detection of
substantial differences between trypanosome and Leishmania DNA
topoisomerase IB with respect to their homologues in mammals has provided a new
lead in the study of the structural determinants that can be effectively
targeted (Reguera et al., 2008).
10. Protein
Kinase
Protein kinases (PKs) are important regulators of many
different cellular processes such as transcriptional control, cell cycle progression
and differentiation, and have drawn much attention as potential drug targets to
treat a wide range of diseases and syndromes, such as cancer, cardiovascular
disease and Alzheimer's disease. The majority of the eukaryotic PKs reside in
clusters of orthologous groups, showing synteny within the three genomes of Leishmania,
however, each species also contains distinctive protein kinases. The protein
kinase complement of the trypanosomatids is about 33% larger than S. cerevisiae, but twice that of the malaria
parasite P. falciparum (Ward
et al., 2004)
10a.Cyclin-dependent
kinases
L. major has an additional unique mitotic-like cyclin, CYCA.
Some cyclin-dependent kinases (CDKs) require phosphorylation on a conserved
threonine residue (T loop, T160 in human CDK1) by a cdc2-activating kinase
(CAK). Many trypanosomatid cdc2-related kinases (CRKs) have a conserved T-loop
residue, suggesting that the CRKs might be activated in vivo by a CAK activity
(Parsons et al., 2005)
10b.Map
kinase
Mitogen-activated protein (MAP) kinases are important
regulators of differentiation and cell proliferation in many eukaryotes.
Several reports describe the identification of MAP kinases (MAPKs) and their
activators, the MAPK kinases (MAPKK), from T. brucei and Leishmania (Bengs et al.,
2005) of the three T.
brucei MAP type kinases described, TbECK1 is the most potential drug
target. These are some of the validated drug targets of Leishmania.
There are more avenues to explore and avail the yet unidentified targets from
the vast resource of leishmanial genome for the betterment of human life.
References
Akopyants,
N. S., et al.
S.M. (2001). Mol. Biochem.Parasitol 113: 337-340.
Bakker,
B. M., et al.
(2000). Proc Natl Acad Sci USA 97: 2087-2092.
Barak,
E., et
al. (2005).
Mol Biochem Parasitol 141(99-108).
Basselin
, M. (2000). Mol Biochem Parasitol 109: 37-46.
Bengs,
F., et
al. (2005).
Mol.
Microbiol 55: 1606–1615.
Carmen,
J. (2008). Molecular and Biochemical Parasitology 160(1): 52-59.
Cordeiro,
A. T. et
al. (2004).
Acta Cryst D60: 915-919.
Cordeiro,
A. T., et al..
(2004). Eur J Biochem 271(13): 2765-2772.
Cunningham,
M. L., et al.
(2001). Mol Biochem Parasitol 113: 199-213.
Docampo,
R., et
al. (2001).
Current Drug Targets Infectious Disorders 1: 51-61.
El
Fadili, A., et al.. (2002). Mol Biochem Parasitol 124: 63-71.
El
Fadili, A., et al. (2003). Biochem Pharmacol 66: 999-1008.
Gagnon
D, F. A., et al..
(2006). Mol Biochem Parasitol 150(1): 63-71.
Gomez-puyou,
A., et
al..
(1995). Chemistry and Biology 2: 847-855.
Handman,
E. (2001). Clin.Microbiol.Rev 14: 229-243.
Hanson,
S., et
al. (2005).
J Biol Chem 267: 2350-2359.
Iniesta,
V., et
al. (2001).
J Exp Med 193: 777-784.
Ivens,
A. C., et al.
(2005). Science 309: 436-442.
Kohl,
L., et
al. (1994).
Eur J Biochem 220: 331-338.
Krieger,
S. (2000). Mol Microbiol 35: 542-552.
Krauth-Siegel R.L., et al.. (1999) Parasitol Today
15 :404–409
Knowles,J.R.,
et
al. (1977) Accounts
Chem Res 10:105-111
Lages,
N. F., et al.
(2007). BMC Systems Biology I (suppl I ):S8.
López,
C., et
al. (2002).
FEBS Journal 269: 3978-3789.
Lorente,
S. O. (2005). Bioorganic and Medicinal Chemistry 13(10): 3519-3529.
Majumdar,
H.K., et
al. (2006) Molecular
Microbiology 62(4): 917-927.
Michels,
P. A. M., et al..
(2000). Parasitol Today 16: 482-489.
Murray,
H. W. (2000). Int J Infect Dis 4: 158-177.
Murray,
H. W., et al.(2005).
Lancet 366: 1561-1577.
Montalvetti A,
et
al. (2003).
J Biol Chem 278(19):17075-83
Montalvetti,A.,
et
al..(2000 )
Biochem J 349:27-34
Opperdoes,
F. R., et al.
(2007). Trends in Parasilol 23: 149-158.
Ouellette,
M., et
al. (2002).
Int J Parasitol 35: 385-398.
Pabón,
M., et
al. (2007).
Parasitology Research 100: 803-810.
Parsons,
M., et
al. (2005). BMC Genomics 6(1): 127.
Peña-Díaz,
J., et
al. .(1997).
Biochem J 324: 619-626
Peacock,C.S.,et al. (2007). Nature Genetics
39:839-847
Reguera,
R. M., et al.
(2008). Curr Drug Targets. 9(11): 966-978.
Richard,
D., et
al. (2002).
J Biol Chem 277: 29460–29467.
Richard,
D., et
al. (2004).
J Biol Chem 279: 54494–54501.
Roberts,
C. W. (2003). Mol Biochem Parasitol 126(2): 129-142.
Roberts
S. C., et al.
(2004). J Biol Chem 279: 23668-23678.
Roberts,
S. C., et al.
(2001). Mol Biochem Parasitol 115: 217-226.
Roberts,
S. C., et al..
(2002). J Biol Chem 277: 5902-5909
Rochette,
A., et
al..(2008) BMC
Genomics 9:255
Rodrigues,
J. C. (2005). Exp Parasitol 111(4): 230-238.
Schmidt.A.,
et
al..
(2002). Curr Top Med Chem 2: 1239–1259.
Seifert
.K., et
al..
(2003). Int J Antimicrob Agents 22: 380-387.
Silva,
M. S., et al.
(2005). FEBS journal 272: 2388-2398.
Singh,
S., et
al. (2007).
Antimicrob agents Chemother 51: 528-534.
Sundar,
S., et
al. (2000).
Clin Infect Dis 31: 1104-1107.
Umasankar,
P. K., et al.
(2005). J Parasitol 91(6): 1504-1509.
Urbina,
J. A. (2002). Molecular and Biochemical Parasitology 125(35-45).
Urbina, J.A. (1997). Parasitology 114 suppl S:91-99.
Verlinde, C.L., et al..(2001) Drug
Resist Updat 4(1):50-65
Vatsyayan,
R., et
al..
(2007). Protein Expr Purif 52(2): 433-440.
Roy,
U., et
al. .L.donovani
Squalene Synthase Accession no. AM229310 www.ncbi.nlm.nih.gov/Genbank
Roy,U.,
et
al.
L.donovani
Arginase Accession no. DQ649412 www.ncbi.nlm.nih.gov/Genbank
Ward,
P., et
al..
(2004). BMC Genomics 5: 79.
Approaches Towards Drug
Development For Leishmniasis: A Review
Suman Gupta* and Shraddha A. Sane
Division
of Parasitology, Central Drug Research Institute,
1. Introduction
Leishmaniasis is a vector-borne disease, which is caused by
obligate intracellular protozoan parasites of the genus Leishmania. This
disease is a severe public health problem in tropical and subtropical regions
of the world. Major characteristic of this disease is its diversity and
complexity (Herwaldt, 1999). More than 20 species of Leishmania cause
leishmaniasis and it is transmitted to humans by ~30 different species of
phlebotomine sandflies (Pearson et al.,
1996). Leishmaniasis is classified as one of the ‘‘most neglected diseases’’ (Yamey
& Torreele, 2002) based on the limited resources invested in diagnosis, treatment,
and control, and its strong association with poverty (Alvar et al., 2006a). The disease is second in mortality and fourth in morbidity among
all tropical diseases (Bern et al, 2008).
Leishmaniasis has several diverse clinical manifestations: ulcerative
skin lesions (CL), destructive mucosal inflammation (MCL), and disseminated
visceral infection (VL), each presenting distinct diagnostic challenges, most
requiring prolonged, expensive drug therapy and each contributing differently
to disease burden. Post kala azar dermal leishmaniasis (PKDL) is characterized
by macular, maculo papular or nodular rash and is a complication of VL that is
frequently observed after treatment. Interactions with malnutrition and HIV
alter the clinical course, and complicate therapeutic strategies. In the
absence of highly active antiretroviral therapy (HAART), the relapse rate after
treatment approaches 100%. Other complicated forms include disseminated
cutaneous leishmaniasis (DCL), diffuse nodular non-ulcerating disease, and
leishmaniasis recidivans (LB), localized slowly progressive non-healing
lesions. Both are rare, difficult to treat, and can be severe. Among these VL is the most severe. It
is caused by L. donovani in the Indian subcontinent,
The recommended drugs for VL & CL, the antimonials, were
first introduced 75 years ago (Deps et al., 2000) however, lack of
response to pentavalent antimonials actively wide spread in India and Sudan led
to the use of Amphotericin –B or Pentamidine. The advances in chemotherapy have
been significant and the concept of choice for treatment in VL is now real. It
is unlikely that
Corresponding author, E-mail: [email protected] and [email protected], Phone: 0522-2612411-18,
Fax: 0522-2623938, 2623405
one single drug or drug
formulation will be effective against all forms of leishmaniasis since (a) the
visceral and cutaneous sites of infections impose varying pharmacokinetic
requirements on the drugs to be used and (b) there is an intrinsic variation in
drug sensitivity of the 20 Leishmania species known to infect humans. In
addition, there are other new problems to be surmounted by novel treatments, namely:
(i) the need for drugs for treatment of VL in Bihar State, India, where there
is acquired resistance to the pentavalent antimonials and (ii) the need for
treatment for VL and CL in immunosuppressed paitents, in particular due to HIV
co - infection, where there is exacerbation of disease or emergence from latent
infection due to the depleted immune response. In the latter case standard
chemotherapy is frequently unsuccessful. (Alvar et al., 2006b).
Among the new drugs discovered, miltefosine, a
hexadecylphosphocholine, is the first promising oral drug which can be used
against leishmaniasis. Other drugs such as paromomycin, sitamaquine, azoles and
azythromycin have been reported as having variable cure rates .Consequently
there is still a real need for new active compounds that can provide
therapeutic benefits but with fewer side effects (Pape, 2008).
2. History
In the 19th century, devastating outbreaks of a chronic
progressive febrile illness with cachexia, hepatosplenomegaly, and high
fatality rates were reported in
3. Epidemiology and Ecology
According to the World Health Organization (WHO) leishmaniasis
is now endemic in 88 countries (16 developed countries, 72 developing countries)
of 5 continents – Africa, Asia, Europe, North- America and South-
America-affecting an estimated 12-14 million people with roughly 1.5-2 million
new cases per year and a total of 350 million people at risk. It has been
estimated that there are 500,000 new cases of VL and more than 50,000 deaths
from the disease each year (only less than that of malaria). Both figures are
approximations as VL is frequently not recognized or not reported. Majority
(more than 90%) of cases occur in 6 countries- Bangladesh, India, Nepal, Sudan,
Ethiopia and Brazil (Croft et al., 2006b).
In
Transmission of leishmaniasis to humans occurs through
sylvatic, domestic, and peridomestic cycles. The distribution is dynamic:
4. Transmission
The transmission of leishmaniasis occurs through Female
Phlebotomine sandflies (Phlebotomus genus in the old world and Lutzomyia
in the new world). They seek a blood meal at or after dusk, becoming infected
if they suck the blood of infected human beings (anthroponoses) or terrestrial
mammals (zoonoses). The life-cycle has two distinct forms; a promastigote
flagellar form found in the gut of the arthropod vector and an amastigote form,
which develops intracellularly in the mammalian host. The sand fly transmits
the disease by inoculation of the promastigote form into the skin. The
parasites are internalized by dendritic cells and macrophages in the dermis and
transform into amastigotes by losing their flagella. They multiply and survive
in phagolysosomes through a complex parasite host interaction. The parasites
disseminate through the lymphatic and vascular systems and infect other
monocytes and the macrophages in the reticuloendothelial system, resulting in
the infiltration of bone marrow, hepatosplenomegaly and sometimes in the
enlarged lymph nodes (lymphadenopathy) (Chappuis et al., 2007).
About 70 of around 1000 known sandfly species transmit
leishmaniasis. Vector competence in most species seems to be controlled by
parasite ability to resist proteolytic enzymes during bloodmeal digestion and
avoid excretion by binding to midgut epithelium. Binding is mediated by
promastigote surface lipophosphoglycan and the phosphoglycan domains differ
between species (Sacks, 2001). Sandfly saliva affects local host immune
responses, promoting experimental cutaneous infection (Sacks &
Noben-Trauth, 2002).
This present review will focus on the drugs currently
available and those which are included in clinical trials, their mode of
action, the experimental models and drug screening procedures with special
emphasis on Visceral Leishmaniasis.
5. Conventional Therapy
Against Visceral Leishmaniasis
5a. Parenterally Effective Agents
Pentavalent
antimonials
N-methylglucamine antimoniate (Glucantime) and sodium
stibogluconate (Pentostam) have been used as a first line of treatment for VL
since the 1940s. Antimony remains the therapeutic cornerstone in all regions
except two: Bihar State, India (houses around 90% of India’s and about 45% of
the world’s cases) where the current approximate 35% cure response has ended
the usefulness of antimony and southern Europe (Sundar et al., 2000)
Relapse rates are less than 5%, but secondary resistance is likely in patients
who relapse unless they are re-treated thoroughly. Effective doses of Sodium
stibogluconate and meglumine antimoniate are 20 mg/kg/day up to a maximum
1275mg over 20 or 30 days given intramuscularly. The maximal tolerated dose is
about 30 mg/kg/day; children tolerate these drugs better than adults. Its
intracellular reduced trivalent form is the active derivative that comes about
through the alteration in parasite bioenergetic pathways and trypanthione
inhibition (Ephros et al., 1999; Wyllie et al., 2004).
Antimonials are toxic drugs with frequent, sometimes life
threatening adverse side effects, including cardiac arrhythmia and acute
pancreatitis. Patients under the age of 2 or aged 45 or over with signs of
advanced disease and /or severe malnutrition are at higher risk of death during
antimonial therapy owing to drug toxicity, slowness of drug action, VL
complications or a combination of these factors (Chappuis et al., 2007).
Pentamidine
isothionate
Pentamidine, an aromatic diamidine has been previously used as
a second line of treatment for VL but its precise mode of action has yet to be
elucidated. Since, it is a competitive inhibitor of arginine transport and noncompetitively
inhibits putrescine and spermidine, its leishmanicidal activity is possibly
mediated via its influence on polyamine biosynthesis and the mitochondrial
membrane potential.Pentamidine was initially proven to be useful in Sbv
resistant kala-azar cases in India but the limiting factors were the expense
and above all the unacceptable toxicity as it causes irreversible insulin
dependent diabetes mellitus and death. Further, it’s declining efficacy (as
only about 70% patients could be cured), has led to its being totally abandoned
in
Amphotericin-B
& its formulations
Conventional Amphotericin B (fungizone®) is a macrolide
polyene, charachterized by hydrophilic polyhydroxyl and hydrophobic polyene
aspects. It is a powerful antileishmanial agent and is a first-line drug in
Liposomal Amphotericin
B
The liposomal amphotericin B formulation, AmBisome®, is
registered treatment for visceral leishmaniasis (Meyerhoff, 1999), but use in
VL endemic regions is limited by cost (US$2,800 per treatment).With recent
preferential pricing offered by the manufacturer to patients in the public
sector in East Africa, it is possible that AmBisome® could become economically
feasible for treatment, even in resource – poor countries (DNDi Annual report
2007- 2008).
Other
commercial Amphotericin B
Lipid formulations have also been manufactured, namely an
amphotericin B lipid complex (Abelcet®) and an amphotericin B colloidal
dispersion (Amphocil™) but their use against VL has not been as extensive as
AmBisome® and they too, are costly. Other re-formulations of Amphotericin B
formulations have been investigated against experimental VL but none have
reached clinical development to date. Approaches to reduce cost include: (i)
efficacy trials of single dose AmBisome treatment for VL, with 90 per cent cure
rate reported to date, and (ii) the use of cheaper liposomal formulations,
already tried for VL (Croft et al., 2006a).
Alternative amphotericin B formulations have been developed to
reduce toxicity and improve drug effect. For example, arabinogalactan
derivatives, nanoparticles and other lipid formulations, or chemical
derivatives, have proved effective in experimental models (Croft et al.,
2006a). The advantages of this association described by these authors include
its physical and chemical stability when lyophilized or soluble, the easy
sterilization by filtration, the drug release profile in the circulation and,
consequently, good elimination by the organism, in addition to the possibility
of i.v. or s.c. administration.A modified meta acrylic polymer of Amphotericin
B has shown promise in experimental work carried out by the Imperial college
team, London in 2007 which includes establishment of adequate efficacy in an in vivo model , size of the polymer, the
ratio of the polymer to Amphotericin B and the actual dose of Amphotericin B.
It is planned by DNDi to advance the most promising Amphotericin B based
formulation by early 2009 (DNDi Annual report 2007- 2008).
Paromomycin
Paromomycin (formerly known as aminosidine) an aminoglycoside
recently registered in
5b. Orally Effective Agents
Miltefosine
Miltefosine, (initially developed as anticancer drug) the
first effective oral treatment for visceral leishmaniasis, including for
antimony-resistant infection, opened the door to self-administered outpatient
therapy. Miltefosine is licensed for visceral leishmaniasis in several regions
and for cutaneous leishmaniasis in some countries. Its rapid development in
Concerns have been expressed about miltefosine’s cost as well
as how to protect the high-level efficacy of this valuable agent from the
effects of poor outpatient compliance and the potential development of
resistance as this drug has a long half life (~150 hours) and parasite
resistance is easily induced in vitro (Perez-Victoria et al., 2006).
Some researchers have suggested combining miltefosine with a second agent in
part to maintain its effect but also reflecting a growing interest in
combination treatments for visceral leishmaniasis.
Sitamaquine
Sitamaquine, an orally active 8-aminoquinoline analog
(8-aminoquinoline (8-[6-(diethylamino)hexyl]amino]-6-methoxy-4-methylquinoline),
was originally developed as WR6026 by the Walter Reed Army Institute in
collaboration with Glaxo Smith Kline in response to a pressing need for orally
effective agents for VL, its effectiveness was validated in animal models.
Several small phase I or II clinical trials have been undertaken with limited
success. The cure rate for VL with sitamaquine in a Kenyan phase II study at a
dose of 1 mg/kg/day for 28 days were 50 percent. Several years later, in a
Brazilian phase II trial, the same dose of sitamaquine cured none of the four
VL patients while a 2 mg/kg/day for 4 wk gave a maximum efficacy of 67 per
cent; surprisingly, a linear correlation could not be sustained as increasing
the dose to 2.5 mg/kg/day resulted in decreased efficacy concomitant with
enhanced adverse effects such as nephropathy and methaemoglobinaemia. In a
multicenter phase II trial in
5c. Other Oral Compounds
Azoles
The last
example of development in new anti-infectious drugs is therapeutic swiching
also called “piggy-back therapy”. Azoles (Ketoconazole, fluconazole, itraconazole,
etc.) are essentially sterol bio-synthesis inhibitors and their efficacy
against L. tropica was first reported by Berman in 1981. Azoles
specifically block ergosterol synthesis and as the presence of ergosterol as a
membrane component is shared between fungi and Leishmania, it accounts
for many antifungal sterol biosynthesis inhibitors (SBIs) to also be
leishmanicidal. Most SBIs impair the biosynthesis of ergosterol by blocking14-á-demethylase,
leading to the accumulation of 14-á-methylsterols. Azoles have been
shown to be active against a wide range of promastigotes and amastigotes. Leishmania
species differ in their sensitivity to azoles as L. donovani, L.
braziliensis and L. amazonensis promastigotes are more sensitive
than L. aethiopica, L. major, L. tropica and L.
mexicana. However, this analogy cannot be extrapolated to clinical studies.
Both ketoconazole and fluconazole have undergone evaluation in VL in
Buparvaquone
Buparvaquone (BPQ) is a hydroxynaphthoquinone and
marketed as Butalex® closely related to a well-known anti- infective
drug, atovaquone. BPQ has been used as an i.m. injection for the treatment
of theileriosis in cattle. For the first time Croft et al (1992) has
tested BPQ against L. donovani infected BALB/c and observed a 62%
suppression of hepatic amastigote burden. Researchers are looking forward to
this drug as a promising antileishmanial agent as it has several
physicochemical properties suitable for topical delivery (low molecular weight,
low melting point, etc.). Attempts have been made to increase aqueous
solubility and absorption, and in this context two phoaphate prodrugs have been
found to show potential in in vitro
& in vivo antileishmanial activity against both visceral and cutaneous
leishmaniasis (Garnier et al., 2007;
Ma¨ntyla et al., 2004).
In 2007, DNDi-
commissioned work by partners at the Universiti Sains
5d. Immunomodulators
Leishmania infection progresses to kala-azar in individuals
who fail to initiate Th 1 response (mediated by IL-2 and IFN-γ). Skewing
of T helper cells towards a Th1 response is considered as a promising
therapeutic strategy. Interferon- γ is one of the principal activators of
macrophages. Clinical trials with IFN- γ alone and/or in conjunction with
Sbv were undertaken. With Sbv it was reported to be
useful in treating severe or Sbv refractory VL in Brazil, however,
in India in a large (n=156) randomized study comparing Sbv alone
with Sbv plus IFN-γ for 15 or 30 days had disappointing results
as the final cure rate with Sbv plus IFN-γ was 42 and 49 per
cent, respectively (Sundar & Chatterjee, 2006).
5e. Drug Combination Strategies
Combination therapy has more potential advantages which
include delay or prevention of the development of resistance (Croft, 2004) and
shorter treatment regimens that could improve compliance and reduce cost.
Unrestricted use of standard antimonials have already posed potential problem
of resistance (Sundar, 2001) so precautionary measures should be taken in case
of monotherapy of arising drugs like miltefosine and paromomycin. Despite of
remarkable work done on combination therapy for Leishmaniasis (Chunge et al.,
1985; Murray & Hariprasad, 1996), it has not yet been adapted as standard
treatment. Limitation is unavailability of effective antileishmanial drug.
Previous studies on the interaction of miltefosine and sodium stibogluconate
have shown synergism in vitro but showed no potentiation in vivo.
Conversely, published reports on the combination of miltefosine with
amphotericin B and miltefosine with paromomycin have shown enhanced efficacy in
vivo in mice model (Seifert
and Croft, 2006). Currently, clinical trials on combination therapy using
paromomycin and miltefosine, AmBisome, miltefosine and paromomycin are being
carried out in
6. Experimental Models in Use in the Drug
Discovery
We will focus here on the specific in vitro and in
vivo assays required in the drug discovery process for Visceral
Leishmaniasis.
6a. In vitro
Assays
Leishmania parasite can be grown in vitro as
promastigotes and amastigotes in axenic conditions. Both these stages have been
exploited for development of primary drug screening procedures.
(i)
Promastigotes:
Drug activity against this extracellular stage is easy to
determine. However, there are significant differences between promastigotes and
amastigotes in biochemistry and sensitivity to standard and experimental drugs
(Croft et al., 2006a). Promastigotes assays are useful cytotoxicity
indicators in bioassay-guided fractionation of plant products. A direct
comparison of the drug susceptibility towards standard antileishmnial drugs,
between amastigotes and axenic amastigotes, demonstrates that the latter
express specific susceptibility to many if, not all the drug tested and
indicates that promastigotes may not be as relevant as axenic amastigotes for
drug screening purpose (Sereno et al., 2007).
(ii) Axenic
Amastigotes:
Screening against axenic amastigotes presents several
advantages; (1) the test is directed against the relevant stage of parasite,
(2) this stage is as easy to manipulate as the promastigote model, (3)
quantification of drug activity is simple and often inexpensive. This can be
achieved by using a cell counter, evaluating the viability of cell population
with a MTT based method, determining ornithine decarboxylase activity or using
a fluorescent dye like Propidium Iodide (PI) and
fluorescence-activated-cell-sorter (FACS). Since, past few years many Leishmania
parasites expressing reporter genes have been selected and the capacity of some
of them to be used in axenic amastigote drug screening protocol has been
assessed (Sereno et al., 2005; Vergnes et al., 2005).The
disadvantages are (i) assay neither test for penetration of compounds into the
host cell nor for activity in the macrophage phagolysosome, (ii) not true
amastigotes (metabolome etc.), (iii) have different metabolic processes than
intracellular amastigotes, (iv) problem of clumping etc.
Ideally to be efficient and exhaustive, a drug screening
procedure requires conditions that tightly mimic the environment encountered by
the target cell. In case of Leishmania, intracellular form of the parasite (amastigotes) represent
the ideal conditions since, this system involves the role played by the host
cell on drug mediated toxicity.
(iii)
Screening Against Intracellular Amastigotes:
The most widely used models for testing drugs against Leishmania
species have involved either murine peritoneal macrophages (J-774) or
human-monocyte tranformed macrophages (THP-1, U937, HL-60) as host cells. These
models show species/strain variation in drug sensitivity (Escobar et al.,
2002; Yardley and Croft, 2005). In these differentiated non-dividing
macrophages, the rate of amastigote division in host cells and drug activity
can be clearly assessed. The activity of test drug is measured by either
microscopical counting of percentage of infected cells or number of amastigotes
/macrophage or colorimetric or fluorometric methods (Neal and Croft, 1984). The
slow rate of division of L. donovani and L. infantum amastigotes
in this model is a limitation. Assays that use dividing host cells must ensure
that the confounding effects of drug activity on both parasite and host cell
number are considered (Croft et al., 2006, a). Many, if not all
classical Screening methods are labour intensive and could not support
automation.
a.
Microscopical Method: In direct
counting assays, drug activity are assessed towards intracellular amastigotes
after Giemsa staining on chamber slide (Sereno et al, 2007). It is
followed by evaluation of drug activity microscopically by determining the
percentage of infected cells as well as the number of amastigotes per cell
through examination of 100–300 macrophages. Counting cells is time consuming
and may give inaccurate determination of IC50, since determination
of the parasite viability through a staining procedure is difficult.
b. Reporter
Gene Assays: Over the past few
years, many Leishmania parasites
expressing reporter genes have been selected and used for testing drug
activity. The main advantage of this technology is its rapidness and
accuracy.
The term reporter gene is used to define a gene with a readily
measurable phenotype that can be distinguished easily over a background of
endogenous proteins. The use of such genes like the firefly luciferase (Ashutosh
et al, 2005), β-galactosidase, chloramphenicol acetyltransferase
(CAT), alkaline phosphatase or the green fluorescent protein (GFP) gene could
considerably facilitate the screening of antimicrobial agents (Naylor, 1999).
The reporter gene technology is generally more sensitive than the other
previously mentioned classical methods. Moreover, reporter proteins bear or
produce an easily detectable response that can be quantified even in
intracellular conditions. In general, methods based on fluorescent proteins are
less sensitive than methods using catalytic reporter genes like luciferase,
β-galactosidase, and β-lactamase. A panel of recombinant parasites
carrying a reporter gene either as an episomal copy or after its integration in
a defined locus, generally the rDNA locus, is currently available.
Various strains of parasites expressing luciferase were
recently developed and their susceptibility towards standard antileishmanial
agents investigated (Roy et al., 2000, Sereno et al., 2001; Ashutosh
et al, 2005). Drug discovery facilities at Central Drug Research
Institute (CDRI), Lucknow have developed Leishmnia
donovani cell lines expressing firefly luciferase reporter gene (luc.) as a
part of episomal vector and established suitability of these cell lines for in vitro screening of antileishmanial
agents (Ashutosh et al, 2005).This system has been adapted to evaluate
compounds in a 96 well microplate format and is being employed (Sundru et al., 2006; Pandey et al., 2007;Gupta et al., 2007) for primary screening of novel synthetic compounds
(Inhouse) and marine extracts (MoES
project) and also for optimization of leads under DNDi supported consortium.
c.
Limitations of Reporter Gene Assays: Reporter genes present several important limitations. Among them the
antibiotic resistance allowing the selection of recombinant parasites could confer
cross-resistance. Neomycin confers resistance towards paromomycin, a lead
candidate drug supported by the Gates foundation. The development of method to
create defined mutants lacking selectable markers could help to overcome this
problem. The way by which the reporter gene is introduced could also have an
impact on the screening. If, the reporters are part of plasmids, the relative
output of reporter may depend on the copy number of the transfected plasmid
(which vary from cell to cell) rather than on the activity of the drug.
Secondly, transforming parasites could have biological consequences either by
disrupting the genomic architecture or just by the presence of the foreign
reporter protein. Thirdly, as previously mentioned for the β-galactosidase
technology, the reporter could have some limitations (i.e. sensitivity,
background activity from host macrophages) making it inaccurate for an in
vitro determination of drug activity against intracellular amastigotes
(Sereno et al., 2007).
d. Multiplexing:
A versatile methodology that
allows for multiple quantifications of drug toxicity against both the host
cells and the intracellular amastigotes could represent a useful tool in the
field of parasite pharmacology. To achieve this goal, reporters must use distinguishable
signal from each other and compatible chemistries, like fluorophores emitting
different wavelengths. Currently, there have been a growing number of examples
using luminescence for multiplexing either in combination with: (i) other
luminescent signals, (ii) fluorescence or (iii) β -galactose assay. Such
methods could also help to directly compare experiments since the results are
expressed as a ratio of the output signal emitted by the host cell on the one
emitted by parasites (Grover et al., 2003; Young et al., 2004).
The usefulness of these approaches for drug screening has to be evaluated on
intracellular parasites like Leishmania.
6b. In vivo Assays
Animal models are expected to mimic the pathological features
and immunological responses observed in humans when exposed to a variety of Leishmania
spp. with different pathogenic characteristics. Many experimental models
have been developed, each with specific features, but none accurately
reproduces what happens in humans. For in vivo testing of new compounds
several animal species have served as experimental host for VL. Important among
them are BALB/c mice and Syrian golden hamster (primary tests), dogs (secondary
tests) and monkeys viz., squirrel, vervet and Indian langur monkeys as
tertiary screens. Animal models enable drug activity to be determined in
relation to absorption (route of administration), distribution (different sites
of infection), metabolism (pro-drugs, immunomodulators), and excretion and to
give an early indication of the toxicity. A suitable laboratory host for the
target parasite (L. donovani) is very important from the point of view
of conducting research on various aspects including host-parasite interactions,
pathogenesis, biochemical changes, prophylaxis, and maintenance of parasites
and above all evaluation of antileishmanial action of newer compounds for
development of new drugs.
Mouse Model
Mostly mice are being used as model for screening of new
compounds, where a relatively low amount of compound is required, which are
available as SPF and inbred strains enabling reproducible results with five
animals per group. Mice are susceptible to most strains and species of Leishmania
in both non-cure and self cure models. The aim of using the animal model is
to find a drug that can be administered orally, be effective in a short course
(< 10 days) and have no indication of toxicity at the highest doses tested
(100 mg/kg). For visceral leishmaniasis inbred strains of mice are widely used
with susceptible, resistant and intermediate strains. The BALB/c mouse is a
commonly used strain, at 18- 20 g, with highly reproducible levels of infection
when an amastigote inoculum is administered i.v. An assay in week two after
infection examines the activity of the drug against the liver infection but not
the spleen infection. The infection in each mouse strain needs to be
characterized for each parasite strain used to ensure that drugs are tested
appropriately. Athymic and scid mice provide a model for treatment of VL
in immunosuppressed cases (Croft et al., 2006a). Hamster
Model
Although many hamster species are susceptible to L.
donovani infection (Smyly & Young, 1924) , the Syrian golden hamster (Mesocricetus
auratus) establishes a good model for VL and provides a more synchronous infection
in the liver and spleen that can develop into a chronic non-cure
infection more similar to human VL (Farrell, 1976; Gifawesen & Farrell,
1989; Hommel et al., 1995). Gupta & Tiwari (2000) have reported the
suitaibility and susceptibility of inbred hamsters in terms of parasite
establishment and longer survival period as compaired to outbred hamsters.Very
recently, Dea-Ayuela et al., (2007)
have studied its suitability and established suitable immunobiological
parameters for in vivo testing of new antileishmanial compounds in the
golden hamster model of visceral leishmaniasis. The clinicopathological
features of the hamster model of VL closely mimic active human disease.
Systemic infection of the hamster with L. donovani results in a
relentless increase in visceral parasite burden, progressive cachexia,
hepatosplenomegaly, pancytopenia, hyper-gamma-globulinemia, and ultimately
death (Gifawesen & Farrell, 1989). Biggest advantage is that biopsy is
possible to monitor pre- & post treatment infection status and all
antileishmanials are active against liver as well as spleen parasites.
A problem in all the models is the determination of drug
activity upon necropsy or biopsy which has been dependent on microscopy to
determine the level of infection. This is now being replaced by quantitative whole
animal non-invasive imaging for parasites. Reporter genes have proved to be an
excellent and promising tool for the detection of parasite stages in target
tissues of animal hosts (Roy et al.,
2000; Lang et al., 2005).
7. Conclusion
New treatments for visceral leishmaniasis have been introduced
and others are undergoing clinical trials. The recent availability of oral
miltefosine for VL has been the most significant development in the past few
years. Care needs to be taken that resistance to these drugs does not develop
and efficiency and safety of drug combinations in greater depth should be
considered. Importantly, the cost of the treatment should be minimized to allow
its dissemination and use mainly in poorer countries, where there is a high
incidence of this disease. Efforts to find new leads and to select new targets
will also contribute to the fight against leishmaniasis and the preparation of
additional resources for the drug discovery pipeline.
References
Alvar,J.,Yactayo,S.,
Alvar,J., Croft,S., Olliaro,P. Adv.
Parasitol. 2006b; 61: 224-274.
Ashutosh,
Gupta, S., Ramesh, Sundar, S. and Goyal, N., Antimicrob. Agent’s chemther.
2005 ; 49: 3776–3783.
Bhattacharya,
S. K. et al. J. Infect. Dis. 2007; 196, 591–598.
Chappuis,
F. et al., 2007. Nat Rev Microb.
2007; 5(11): 873-882.
Chunge,
C. N. et al., Trans. R. Soc. Trop. Med. Hyg. 1985; 79: 715–718.
Collin, S., Davidson, R., Ritmeijer, K., Keus, K.,
Melaku, Y., et al. Clin Infect Dis. 2004; 38: 612–619.
Costa, C.H. et
al., Amm. J.Trop. Med. Hyg. 2002; 66: 334-337.
Croft SL, Hogg J, Gutteridge
WE, et al. J Antimicrob Chemother
1992 ;30 :827–32
Croft,
S. L. 2004. WHO/TDR Scientifi Working Group Report on Leishmaniasis TDR/SWG/04.
World Health Organization,
Croft,
S.L., Seifert, K., Yardley, V.,
Croft
S.L., Sundar S., Failamb A.H., Clin Mocrob Rev 2006b ; 19 :
111-126.
Davidson,
R.N., Medicine 2005; 33:8.
Dea-Ayuela,
M.A., et al. Vet Res Commun 2007; 31: 703-717.
den Boer, M. & Davidson, R. N. Expert Rev.
Anti Infect. Ther.
2006; 4 :187–197.
Deps, P.D. et al .Rev Soc Bras Med Trop
2000 ;33:535-543.
Desjeux,
P., Public health aspects and control. Clin Dermatol 1996; 14: 417–423.
DNDi
Annual Report 2007-2008.
Ephros,
M. et al, Antimicrob. Agents chemther.1999; 43: 278-282.
Escobar,
P. Matu, S., Marques, C., Croft, S.L., Acta Trop. 2002; 81:151-157
Farrell, J.P.,Exp Parasitol .1976 ; 40 :
89-94.
Garnier, T.,Mantyla,A.,Jarvinen,T. et
al., J. Antimicrob. Chemothr. 2007 ;60(4) : 802-810.
Gifawesen,C. & Farrell, J.P.,Infect Immun. 1989;
57 : 3091-3096.
Grover, G.S., et al, J. Biomol Screen 2003;
8:239-246.
Gupta, L. et.al.Bioorg.Med.Chem.Lett.2007; 17
: 4075-4079.
Gupta, S. & Tiwari, S., J. parasit. Dis. 2000;
24 : 211-213.
Gupta,
S., Tiwari, S., Bagchi, A.K. and Katiyar, J.C. Curr. Sci. 1997;
73: 456-459.
Herwaldt,
B. L., Leishmaniasis. Lancet. 1999; 354:1191–1199.
Hommel, M., et al., Ann
Trop Med Parasitol 1995; 89 : Suppl 1, 55-73.
Kotra
L., Haddad, J., Mobashery, S. Antimicrob. Agents chemther.2000; 44 : 3249-3256.
Lang,T.
et al. ,Cell Microbiol. 2005 ; 7 :
383-392.
Lux,
H. et al , Mol Biochem Parasitol 2000;111:1-14.
Lux, H., Hart D.T., Parker P.J., Klenner T. Adv
Exp Med Biol 1996 ; 416 : 201-211.
Maarouf
et al, Exp Cell Res 1997; 232: 339-348. Ma¨ntyla¨ A, Garnier T,
Rautio J et al. J Med Chem 2004; 47:
188–195.
Murray,
H. W., Berman, J. D., Davies, C. R, Saravia, N. G., Lancet 2005;
366: 1561–1577.
Naylor,
L.H., Biochem Pharmacol 1999; 58:749-757.
Neal,
R.A. and Croft S.L., J Antimicrob Chemother 1984; 14: 463-475
Pagliano,
P., Carannante, N., Rossi, M., Gramiccia, M., Gradoni, L., et al. J Antimicrob Chemother. 2005; 55:
229–233.
Pandey,
S., Suryawanshi, S.N., Nishi, Goyal, N.,Gupta, S.Eur. J. Med. Chem.2007;
42 : 669-674.
Pape,
P.L. J Enz Inhb Med Chem 2008; 23:5,708-718.
Pearson,
R. D. & Sousa, A. Q., Clin. Infect. Dis. 1996; 22: 1–13.
Rey,
L.C., Martins, C.V., Ribeiro H.B.,
Sacks,
D. and Noben-Trauth, N., Nat Rev Immunol 2002; 2: 845–858.
Sacks,
D.L.,Cell Microbiol 2001; 3: 189–196..
Sereno,
D., Roy, G., Lemerse, S., Papadopoulou, B., Ouellette, M., Antimicrob.
Agents chemther. 2001; 45:1168-1173.
Sereno,D,
Alegre, A.M., Silvestre, R., Vergnes, B., Ouaissi, A., Antimicrob. Agents
chemther 2005; 48:808-12.
Sereno,D.
et al Parasit. Int. 2007; 56:3-7.
Seifert
.K. and Croft ,S. L. Antimicrob Agents
Chemother. 2006, 50(1): 73–79.
Smyly,H.J.
and Young,L.W.Proc.Soc.Exp.Biol.Med. 1924; 21,354.
Sundar, S. Trop.Med.
Int. Health. 2001; 6 : 849–854.
Sundar, S. & Chatterjee, M., Indian
J Med Res 2006 ; 123 : 345-352
Sundar,
S. & Murray, H.W. Bull. World Health Organ 2005;
83 : 394–395.
Sundar,
S., Jha, T. K., Thakur, C. P., Sinha, P. K. and. Bhattacharya,
S. K. N. Engl. J. Med. 2007 ; 356 : 2571–2581.
Sundar,
S., More, D.K., Singh, M.K., et al. Clin Infect Dis 2000;
31: 1104–1106.
Sundru,
N., Agarwal, A., Katiyar, S.B., Nishi, Goyal, N., Gupta, S., Chauhan, P.M.S. Bioorg.Med.Chem.Lett.
2006 ; 14 : 7706-7715.
Thakur,
C.P. Trans. Roy. Soc. Trop. Med. Hyg. 1984 ; 78 : 391- 398.
Vergnes, B., Vanhille, L., Ouaissi, A., Sereno, D.
Acta Trop. 2005 ; 94 : 107-115.
Wyllie
S., Cunningham M.L., Fairlamb A.H., J Biol Chem 2004 ; 279 :
39925-39932.
Yardley,V.
and Croft,S.L. In Handbook of animal models of infection.Zak O, editor,
Yardley,V., et al, Am J Trop Med Hyg
2005 ; 73 : 272-275.
Yamey,G.
& Torreele, E., Bristish Medical
Journal 2002 ; 325 : 176-177.
Young, K.H., et al, Methods Enzymol 2004 ; 389 : 277-301.
Mechanisms of Drug Resistance in Kala-azar
Neena Goyal and Ashutosh
Division of Biochemistry,
Central Drug Research Institute,
1. Introduction
Leishmaniasis is a disease complex caused by an obligate
intracellular protozoan parasite of the genus Leishmania, which is transmitted to humans by the bite of female
sandflies of the genus Phlebotomus
(in the
The disease is endemic in 88 countries with approximately 350
million people at risk (TDR, 2005). The
estimated annual incidence of kala-azar is around 5,00,000 in 61 countries with
90% of these cases being confined to 5 countries namely
Due to non-existence of effective vaccine to date,
chemotherapy is the only effective way to control Leishmania infections.
Available drugs are limited in number and each has various shortcomings.
Pentavalent antimony has long been the cornerstone of anti-leishmanial chemotherapy for
more than 60 years and still plays a leading role in treatment, but resistance
to this drug class is so high in some parts of the world (particularly in North
East India) that it is quickly becoming obsolete (Sundar et al., 2000). Second-line drugs include pentamidine and
amphotericin B, but these drugs have not experienced widespread use due to
toxicity and cost. In recent years four new potential therapies namely
liposomized amphotericin B (Meyerhoff, 1999),
oral miltefosine (Sundar et al, 2002),
paromomycin (Thakur et al. 2000; www.iowh.org) and oral sitamaquine (previously WR6026) (Wasunna et al. 2005) (http://science.gsk.com/about/disease.htm)
have been introduced which are under various stages of development.
Due to anthroponotic cycle, intense transmission, poverty,
illiteracy, misuse of the drug and poor health care facilities, resistance to
Sb has been developed. It suggests that resistance could also develop to other
antileishmanial drugs as they are introduced as mono-therapy for long periods.
Further, increasing numbers of HIV/VL-coinfected patients will be a potential
source for emergence of drug resistance because these patients have high
parasite burden, a weak immune response, respond slowly to treatment, high
relapse rate, and could be a reservoir of drug-resistant parasites (Molina et al.
2003). Prevention and circumvention of resistance towards antimonials
has become WHO priority (http:// www.who. int/tdr/diseases/ leish/ strategy.htm).
In this, review is focused on the known or potential resistance mechanisms to
these anti-leishmanials drugs.
2. Resistance Mechanism to
Pentavalent Antimonials
In Leishmania,
antimony resistance is interplay between conversion of pro-drug to active
molecule, uptake, efflux and sequestration of active drug (Croft et al., 2006; Ashutosh et al., 2007) which has been studied in
great details using laboratory mutants mostly of L. tarantolae that are resistant to heavy metal arsenite and cross
resistant to antimony (Rosen, 2002). It is generally agreed that the drug in
clinical use i.e. pentavalent antimony (SbV) is a prodrug that needs to be
converted in to trivalent antimony (SbIII), either non-enzymatically by thiols
(Ferreira Cdos et al., 2003; Frezard et al., 2001) and/or by parasite
specific enzymes named thiol dependent reductase (TDR1) (Denton et al., 2004) and antimonite reductase,
ACR2 (Zhou et al., 2004). Further,
the site of conversion (macrophage or amastigote) is still unclear. One study
has shown that axenically grown amastigotes, but not promastigotes, reduce
Sb(V) to Sb(III) (Shaked-Mishan et al.,
2001), while other studies have suggested that reduction most likely takes
place in the macrophages (Roberts & Rainey, 1993; Sereno et al., 1998). As the macrophages do not
efficiently reduce SbV to SbIII (Wyllie & Fairlamb, 2006), parasites are
not exposed to lethal amount of SbIII within the macrophages, therefore
reduction of pentavalent antimony become critical event and loss of reductive
activity (enzymatic and non-enzymatic) in the parasite could lead to
resistance.
To be active against Leishmania,
antimony has to enter the host cell, cross the phagolysomal membrane and act
against intracellular parasite. In number of species the accumulation of SbV is
higher in axenic amstigotes than in promastigotes (Brochu et al., 2003) while SbIII accumulates almost equally in both
stages. This suggests that two forms of antimony do not enter by the same
route. It was speculated that SbV enter via a protein recognizing a sugar
moiety like structure shared with gluconate (Brochu et al., 2003) while uptake of Sb(III) and As(III) is mediated by
aquaglyceroporins (AQPs) (Gourbal et al., 2004). The expression of AQP1
was found to be down-regulated in antimony-resistant mutants of several Leishmania
species (Marquis et al., 2005) as
well as in Sb(V)-resistant clinical isolates from
In resistance, increased efflux of drug or its active molecule
is very common in various pathogens including Leishmania. This could be achieved either by amplification of the
transporters (Haimeur et al., 1998) or
by increased sequestration of active drug in conjugation with thiols (Oullettee
et al., 2002; Wyllie et al., 2004). Indeed both mechanisms
have been demonstrated in several laboratory resistant mutants (Haimeur et al., 2000; Brouchu et al., 2003; El Fadilli et al., 2005; Marquis et al., 2005). Both classes of ABC transporters
known for MDR in cancer cells i.e. P-glycoprotein (P-gp) and multi drug
resistance related protein (MRP) have been reported to be amplified in various
species in response to different drugs under laboratory conditions (reviewed by
Leandro & Campino, 2003). Two out of eight putative MRP1 homologues in Leishmania genome are known to be
involved in metal resistance (reviewed by Brost & Elferink, 2002). The
first one is PGPA (renamed as MRPA) which has been found to be amplified in a
number of laboratory resistant mutants of Leishmania species and its
role in resistance has been confirmed by transfection studies (reviewed by
Ouellette et al. 2004) . It confers
resistance by sequestration of metal–thiol conjugates (Le´gare´ et al., 2001). Although, MRPA has not
been found to be upregulated in a antimony-resistant L. donovani field
isolates from
The second ABC transporter, PRP1 has been shown to confer
cross-resistance to antimony in laboratory mutant resistant to pentamidine
resistance (Coelho et al., 2003). However,
the mechanism by which it confers resistance remain to be determined.
Another transporter which also plays a role in active efflux
of metal-thiols conjugates in energy dependent manner has been reported in L. tarentolae laboratory mutants (Dey et al., 1994).
It is quite interesting to note that the antimony resistant L.
donovani field isolates also induce the upregulation of MRP and P-gp in macrophages, resulting
in a less accumulation of intracellular Sb following treatment with SAG thus
favoring parasite replication (Basu et
al., 2008).
The laboratory resistant mutants of all Leishmania species exhibited significantly increased level of
intracellular thiols namely cysteine, glutathione (GSH), spermidine and
trypanothione (TSH) suggesting the role of thiols in resistance (reviewd by
Ouellette et al. 2004). This is due
to amplification of γ-GCS (encodes γ-glutamylcysteine synthetase) and
over expression of ODC (encodes
ornithine decarboxylase), the rate limiting enzymes involved in synthesis of
GSH and spermidine respectively and amplification of tryparedoxin, an important
enzyme of thiol metabolism (Lin et al 2005) resulting in increased levels of
TSH. Further, lowering of thiols by
inhibiting these enzymes with their specific inhibitors,i.e. l-buthionine-(SR)-sulphoximine
(BSO) and dl-![]() -difluoromethylornithine
(DFMO) resulted in reversal of resistance (Arana et al., 1998; Grondin et al.,
1997). The acquired resistance by co-transfection of ODC or γ-GCS with MRPA in wild-type cells was also
reversed by a thiol depletor (El Fadili et
al. 2005). It was therefore, established that in laboratory mutants MRPA and increased TSH act
synergistically for the resistance to heavy metals (Ouellette
et al., 2004). Interestingly,
role of increased the levels of reduced intracellular thiols (due to increased
expression of trypanothione reductase, γ-GCS
and ODC ) have also been observedin
clinical antimony resistance (Mittal et
al. 2007, Mandal et al. 2007,
Mukherjee et al. 2007), which is also
reversed by BSO both in vitro (Mittal
et al., 2007) and in animal model
(Carter et al., 2005).
-difluoromethylornithine
(DFMO) resulted in reversal of resistance (Arana et al., 1998; Grondin et al.,
1997). The acquired resistance by co-transfection of ODC or γ-GCS with MRPA in wild-type cells was also
reversed by a thiol depletor (El Fadili et
al. 2005). It was therefore, established that in laboratory mutants MRPA and increased TSH act
synergistically for the resistance to heavy metals (Ouellette
et al., 2004). Interestingly,
role of increased the levels of reduced intracellular thiols (due to increased
expression of trypanothione reductase, γ-GCS
and ODC ) have also been observedin
clinical antimony resistance (Mittal et
al. 2007, Mandal et al. 2007,
Mukherjee et al. 2007), which is also
reversed by BSO both in vitro (Mittal
et al., 2007) and in animal model
(Carter et al., 2005).
Recently, antimony-resistant isolates have been shown to
down-regulate the expression of γ
-GCS of macrophages and GSH levels, minimizing the intra-macrophage reduction
of Sb(V) to its toxic form Sb(III) (Carter et
al., 2006; Wyllie & Fairlamb, 2006). Thus, there is clear indication
that the thiols have a dual role in antimony resistance, i.e. sensitization of
the parasite by the reduction of pentavalent to trivalent antimony and
promoting resistance by forming conjugates with trivalent antimony for efflux
and/or sequestration.
Others mechanism includes increase in levels of heat-shock
proteins. Recently, Hsp70 and Hsc70 have been identified in laboratory mutant
by functional cloning (Brochu et al.,
2004). However, transfection of an HSP gene does not confer resistance
directly, but rather increases the tolerance of the cell to metals. Further, it
is still not known how Hsp70 provides resistance and whether this mechanism
operates in field isolates too.
3. Resistance to Pentamidine
Pentamidine isothionate has been used as second-line drugs for
the treatment of SbV refractory patients in 1980s. However, a quick decline in
the response rate from 95% to 70% within a decade period suggested that
parasites were becoming resistant (Sundar, 2001).Pentamidine resistant promastigotes
of L. donovani and L. amazonensis were shown to have reduced
uptake and increased efflux (Basselin et
al., 2002). The specific transporters for pentamidine uptake have been
characterized that may have some role in resistance (Bray et al., 2003). An ABC transporter PRP1 was identified by functional
cloning in mutants selected for pentamidine resistance (Coelho et al., 2003). Interestingly, verapamil,
a calcium-channel blocker was shown to inhibit both pentamidine efflux
(Basselin et al., 2002) and PRP1-mediated
resistance (Coelho et al., 2003).
Future studies are warranted to investigate in more detail the route of
pentamidine entry into mitochondria and the main route of its extrusion outside
the cell.
4. Resistance to
Amphotericin B and its Formulations
Amphotericin B is now a second-line treatment for VL when
antimonial therapy fails. To reduce toxicity AmpB has been reformulated to its
liposomal form, AmBisome®. This is registered treatment for VL and cured 90% of
patients in
Resistance to AmB has been induced in vitro and was found to be associated with DNA amplification
(Singh et al., 2001) although the
link between these amplicons and resistance has yet to be confirmed. A resistant clone of L. donovani promastigotes
showed a significant change in plasma membrane sterol profile, ergosterol being
replaced by a precursor, cholesta-5,7,24-trien-3β-ol (Mbongo et al., 1998) which results in decrease in
the binding affinity of AmB to these sterol-modified membranes. This replacement
probably results from a defect in C-24 transmethylation due to loss of function
of S-adenosyl-L-methionine-C24-Δ-sterol-methyl-transferase (SCMT).
In L. donovani promastigotes two transcripts of the enzyme have now been
characterized, one of which was absent in the amphotericin B-resistant clone,
the other was overexpressed but without a splice leader sequence which would
prevent translation (Pourshafie et al.
2004).
5. Resistance to Miltefosine
Miltefosine is the first effective oral treatment of VL and
the latest antileishmanial drug to enter the market, registered in
In vitro resistance has been developed in promastigote of L.
donovani and possible resistance mechanisms include reduced drug uptake,
differential plasma membrane permeability, faster drug metabolism and efflux of
the drug (Seifert et al 2003). There was >95% reduced accumulation of 14C-labeled
miltefosine in resistant L. donovani parasites while binding of drug to the
promastigote membrane and drug efflux was shown to be similar in sensitive and
resistant lines (Perez-Victoria et al 2003a). Subsequent studies on these cell
lines, described single distinct point mutations in novel plasma membrane
P-type transporter (LDMT gene) from
the aminophospholipid translocase subfamily, responsible for reduced uptake of
the drug (Perez-Victoria et al 2003a & b). Previously, it has been shown
that Leishmania cells selected for resistance to the anticancer agent
daunomycin over expressing the P-glycoprotein gene MDR1 (Perez-Victoria et al., 2001) were cross-resistant to
miltefosine (Perez-Victoria et al.,
2001). It was suggested that MDR1
transports substrates into the secretary compartments that are then extruded
from the parasite by exocytosis (Dodge et
al., 2004). However, the potential relevance of these observations need) to
be extended to miltefosine-resistant amastigotes before clinical implications
can be considered.
6. Resistance to Paromomycin
Paromomycin (PM), an aminoglycoside antibiotic, originally
identified as an antileishmanial in the 1960s is now undergoing phase III
trials in
7.
Resistance to Sitamaquine
Another oral drug that might have an impact on VL is the
8-aminoquinoline derivative sitamaquine (previously WR6026), currently in
development (http://www.gsk.com) stage with varying
levels of success (67-92% cure rate) (Jha et al 2005). Sitamaquine is rapidly
metabolized, forming diethyl and 4-CH2OH derivatives, which might be
responsible for its activity. The activity of sitamaquine metabolites against Leishmania
spp. has not been reported. The mode of action is not known but could
involve “futile redox cycling” as proposed for primaquine. Very little is known
regarding its resistance mechanisms due to limited clinical use and no reported
resistance.
8. Concluding Remark
Few drugs are available for treating Leishmania infections and the emergence of drug resistance is
complicating the control of Leishmaniasis. A better understanding of resistance
mechanisms of known drugs may open new vistas for their rational use and define
combinations, this would help to minimize resistance and to achieve more
effective treatments. Global analysis of resistance is likely to unravel both
primary resistance mechanisms and secondary resistance mutations such as those
involved in fitness compensatory mutations. These efforts are absolutely
required to find new targets and drugs for reversing the recrudescence of
parasitic infections.
References
Arana,
F.E. et al. (1998). Biochem Pharmacol
56, 1201-1208
Ashutosh et al. (2007). J Med
Microbiol. 56: 143-153.
Basselin M. et al. (2002). Antimicrob. Agents Chemother. 46, 3731–3738.
Basu J. M. et al. (2008). Antimicrob Agent Chemother. 52: 1080-1093.
Brochu C. et al. (2003). Antimicrob Agents Chemother 47, 3073–3079.
Brochu C. et al. (2004). Cell Stress Chaperones 9, 294–303.
Brost P. & Elferink R. O.
(2002). Annu Rev Biochem. 71,
537–592.
Carter K. C. et al. (2005). Parasitology. 131, 747–757.
Carter K. C. et al. (2006). Antimicrob Agents Chemother. 50, 88–95.
Coelho A. C. et al. (2003). Mol. Biochem. Parasitol. 130:83–90.
Croft S. L. et al. (2006). Clin Microbiol Rev. 19, 111–126.
Decuypere S. et al. (2005). Antimicrob Agents Chemother. 49, 4616–4621.
Dey S. et al. (1994). Mol Biochem
Parasitol. 67, 49–57.
Dey S. et al. (1996). Proc Natl Acad
Sci
Di Giorgio C. et al. (1999). J. Antimicrob. Chemother. 44:71–76.
Dodge M.A. et al. (2004). Mol. Microbiol. 51, 1563–1575.
El Fadili K. et al. (2005). Antimicrob Agents Chemother. 49, 1988–1993.
Ferreira Cdos s. Et al. (2003). Biometal 16, 441-446.
Fong D. et al. (1994). Am. J. Trop.
Med. Hyg. 51:758–766.
Frezard F. et al. (2001). Antimicrob Agents Chemother 45, 913–916.
Gourbal B. et al. (2004). J Biol Chem. 279, 31010–31017.
Grondin, K. et al. (1997). EMBO J 16, 3057–306.
Guerin P. J. et al.
(2002). Lancet Infect Dis. 2,
494–501.
Haimeur, A.et al.(1998) Antimicrob Agents Chemother 42, 1689-94.
Haimeur, A.et al. (2000). Mol Biochem Parasitol 108, 131–135.
Herwaldt B. L. (1999). Lancet. 354, 1191–1199.
Jha TK et al. (2005). Am J Trop Med Hyg. 73: 1005-11.
Leandro C. & Campino L.
(2003). Int J Antimicrob Agents. 22,
352–357.
Légaré, D (2001). J Biol Chem 276, 26301-26307.
Lin Y. C. et al. (2005). Mol Biochem Parasitol. 142: 66-75.
Maarouf M. et al. (1995). Parasitol Res. 81:421–425.
Maarouf M. et al. (1997). Exp Cell Res. 232:339–348.
Maarouf M. et al. (1998). Parasite. 5:167–173.
Maharjan M. et al. (2007). Am J Trop Med Hyg. 79: 69-71
Mandal, G. et al. (2007). Parasitology. 134: 1679-1687.
Marquis N. et al. (2005).
Mol Microbiol. 57: 1690-1699.
Mbongo N. et al. (1998). Antimicrob Agents Chemother. 42:352–357.
Meyerhoff A. (1999). Clin. Infect. Dis.28, 42-51.
Mittal M. K. et al. (2007). Am J Trop Med Hyg. 76:681-688.
Molina r. Et al. (2005). Am. J. Trop. Med. Hyg. 97, 29-45.
Mukherjee, A. et al. (2007). J Antimicrobiol Chemoth. 59: 204-211.
Mukhopadhyay R. et al. (1996). Proc Natl Acad Sci
Ouellette M. et al. (2002). Mol Med. Parasitol. Academic Press, 395-430.
Ouellette M. et al. (2004). Drug Resist Update 7, 257–266.
Perez-Victoria F. J. et al. (2003a). Antimicrob Agents Chemother 47, 2397–2403.
Perez-Victoria F. J. et al. (2003b). J Biol Chem. 278:49965–49971.
Perez-Victoria J.M. et al. (2001). Antimicrob Agents Chemother. 45, 2468–2474.
Pourshafie M. et al. (2004). Antimicrob Agents Chemother. 48:2409–2414.
Roberts W. L. & Rainey P.
M. (1993). Antimicrob Agents Chemother
37, 1842–1846.
Rosen B. P. (2002). Comp Biochem Physiol A Mol Integr Physiol.
133, 689–693.
Seifert K. et al. (2003). Int J Antimicrob Agents. 22:380–387.
Sereno D. et al. (1998). Antimicrob Agents Chemother 42, 3097–3102.
Shaked-Mishan P. et al. (2001). J Biol Chem. 276, 3971–3976.
Singh A. K. et al. (2001). Exp Parasitol. 99:141–147.
Sundar S. & Murray H. W.
(2005). Bull World Health Organ. 83,
394–395.
Sundar S. (2001). Trop Med Int Health. 6, 849–854.
Sundar S. et al. (2000). Clin Infect Dis. 31, 1104–1106.
Sundar S. et al.(2002). N.Eng.J.Med. 347,
1739-1746.
Sundar S. et al.(2004). Clin Infect Dis. 38, 377-383.
Thakur C.P. et al. (2000). Trans R Soc Trop Med Hyg. 94, 429–431.
Wasunna M. K. Et al (2005) Am. J. Trop. Med. Hyg. 73, 871-876.
Wyllie S. & Fairlamb A.
H. (2006). Biochem Pharmacol. 71,
257–267.
Wyllie S. et al. (2004). J. Bio. Chem. 279, 39925-39932.
Zhou Y. et al. (2004). J Biol Chem.
279, 37445–37451.
Is Vaccination Feasible against Kala-Azar?
Mukesh Samant and Anuradha
Dube
Division of Parasitology, Central Drug Research Institute,
1. Immunization Against Leishmaniasis
The ultimate goal of a
vaccine/immunogen is to develop long-lived immunological protection.
Vaccination leads to enhanced responses that either completely prevent
infection or greatly reduce the severity of the disease. Leishmaniasis in
general, but particularly CL is probably one of a few parasitic diseases that
are most likely to be controlled by vaccines. The relatively uncomplicated
leishmanial life cycle and the fact that recovery from a primary infection
renders the host resistant to subsequent infections indicate that a successful
vaccine is feasible. Therefore, the important step in a rational design of a
vaccine is to understand the immunological correlations of protection.
2. Immune Response of the Host During Active Leishmania Infection
Cellular
Studies of anti-Leishmania vaccine candidates have advanced in recent years due to
the understanding of cell-mediated immunological mechanisms for controlling
infection. Most current knowledge, however, is based on experimental
mouse/hamster models and cannot be extrapolated to dogs or humans. Acquired
resistance to leishmaniasis is mediated by T-cells. CD4+ lymphocytes are
crucial for resistance and CD8+ are more involved in memory than as effector
cells. In humans, a correlation between Th1 cell responses and resistance and
healing of cutaneous leishmaniasis was described with a predominance of cells
producing IFN-γ, while a mixed Th1/Th2 response with IL-4 and IL-10
characterized mucocutaneous and chronic cutaneous lesions. Other studies showed
this mixed cytokine profile in cutaneous leishmaniasis and in natural
resistance, with IL-4 and IL-10 predominating in early infection and a Type-1
immune profile in patients with older lesions, or a main exacerbated Th1
response with high levels of IFN-γ and TNF-α in mucosal patients. In
VL, no association of IL-4 with active disease was described; however sustained
levels of IFN-γ and a direct correlation between increase in IL-10 and
active disease by L. donovani were
detected, while TNF-α was significantly elevated in cases of PDKL. The
mixed cytokine pattern has been confirmed in active disease by L. chagasi.
In the mouse model for VL, Leishmania specific Th2 cells and
antigen presenting cells are involved in suppression of T-cell responses. The
DTH response is suppressed in mice, dogs, and humans and is recovered after
cure. Macrophage mediated suppression leads to the increase in parasite burden
and antigen energy, and is linked to either defective antigen presentation or
inhibition of expression of class I and class II major histocompatibility
complex molecules. Immunosuppression in mice is related to enhanced TGF-β
expression, IL-10, and the possible participation of CD4+ CD25+ regulatory
cells. In VL, apoptosis of T CD4+ cells is accompanied by a decrease in IL-2
and IFN- γ. In addition, apoptosis is detected in inflammatory cells in
liver and spleen during infection with L.
donovani. Soluble factors in serum, such as triglycerides, may also be
involved in immunosuppression. These disappeared if serum is delipidated. The
murine model for CL is considered one of the best for the study of the mechanisms
controlling the T helper Th1 and Th2 cell balance. In the resistant strains
(C57Bl/6, C3H, CBA) resolution of the limited infection is mediated by Th1
cells secreting IFN-γ in response to IL-12. IFN-γ activates the
parasiticide activity of macrophages through the synthesis of inducible nitric
oxide synthase (iNOS), which leads to production of nitrogen radicals. In
contrast, the susceptible Balb/c mice develop a main Th2 response that results
in progression of lesions and systemic disease. In draining lymph nodes, CD4+
cells secrete IL-4.
Infected
Syrian hamster
with L. donovani, reproduced
the clinicopathological features of human VL, and investigation into the
mechanisms of disease in the hamster revealed striking differences from the
murine model. Uncontrolled parasite replication in the hamster liver, spleen,
and bone marrow occurred despite a strong Th1-like cytokine (IL-2, IFN-g, and
TNF/lymphotoxin) response in these organs, suggesting impairment of macrophage
effector function. Indeed, throughout the course of infection, inducible NO
synthase (iNOS, NOS2) mRNA or enzyme activity in liver or spleen tissue was not
detected. The impaired hamster NOS2 expression could not be explained by an
absence of the NOS2 gene, over production of IL-4, defective TNF/lymphotoxin
production (a potent second signal for NOS2 induction), or early dominant
production of the deactivating cytokines IL-10 and TGF-b. Thus, although a Th1-like cytokine response
was prominent, the major antileishmanial effector mechanism that is responsible
for control of infection in mice was absent throughout the course of
progressive VL in the hamster.
Humoral
Human kala-azar is characterized by high
titers of Leishmania-specific
antibodies appearing soon after infection and before the development of
cellular immunological abnormalities. The role of these antibodies in disease
resolution or protection is largely unknown. The Th1 cytokine IFN-γ
probably upregulates IgG1 and IgG3 in humans, while the Th2 cytokines IL-4 and
IL-5 stimulate the production of high levels of IgM, IgE, and IgG isotypes such
as IgG4. Analysis of the Leishmania
specific Ig isotypes and IgG subclasses in VL patient sera revealed elevated
levels of IgG, IgM, IgE and IgG subclasses during disease. Drug resistance was
associated with a reduction in IgG2 and IgG3. A marked elevation of IgG1,
however, was observed in all these patients. A successful cure corresponded
with a decline, most significantly, in the levels of IgE, IgG4, and IgG1.
In hamster model of VL the level of Leishmania-specific
IgG increased dramatically with a detection of specific antileishmanial
antibody in serum. It has been reported that the serum concentration of IgG2
was increased almost 6-fold in the L.
donovani infected hamster compared with uninfected controls. The levels of
IgA and IgG1 was also increased 2- to 3-fold, whereas the IgM and IgG3
concentrations did not increase significantly.
3. Strategies for
Immunization/ Vaccination
The immunity observed after the
therapeutic cure of kala-azar, suggests that vaccination against leishmaniasis is feasible and within the
reach of conventional immunization methods. Several vaccine strategies have
been developed. The “first generation” vaccines composed of killed parasites
with or without adjuvant, are undergoing various stages of phase I (safety), II
(reactivity) and III (efficacy) trials in humans conducted in
An ideal anti-leishmanial
vaccine would need to possess several attributes, but not all of them may be
easily achievable. These include; I) Safety II) Affordability and Stability; III) A good vaccine should stimulate a strong, protective and long
lasting immune response, IV) Effectiveness against species causing CL
and VL, V) Stability at room temperature; VI) A good vaccine should induce the appropriate immune responses-VII)
Effectiveness as a prophylactic as well as a therapeutic vaccine; VIII) A
single dose of vaccine should confer robust, long-lived immunity.
The
human efficacy trials involving the so called ‘‘killed vaccines’’, composed of
crude total parasite antigens, started in
The
most striking aspect of the first-generation human vaccines is that a
leishmanin skin test (LST) is used for candidate selection and for confirmation
of immunogenicity. Whenever the LST is performed, VE is obtained among the
individuals whose skin tested positive
whereas no efficacy is detected in assays that did not include an LST.
One exception is a study done in
Live
This
category includes vaccines made of genetically modified, ‘‘knock-out’’ Leishmania spp., which lack essential
genes, such as dyhydrofolate-reductase thymidilate synthase, cystein-proteinase
or biopterin transporter. These parasites undergo a short life cycle, enough to
generate a specific immune response causing abortive infection and no disease
in man. Another approach is to introduce in the Leishmania genome ‘‘suicidal cassettes’’ including drug sensitive
genes, such as L. major expressing
the ganciclovir-sensitive thymidine kinase gene of Herpes I virus or the Saccharomyces
cerevisae cytosine deaminase gene sensitive to 5-fluorocytosine. The
authors of these studies have suggested that in Iran, where ‘‘leishmanization’’
is used as vaccine challenge to ensure consistency of infection rates and
reduce the number of participants and duration of the assay, the use of parasites
with ‘‘suicidal cassettes’’ would guarantee effective treatment of
non-resolving lesions or of infections resistant to the usual chemotherapy.
However, the use of live challenge for humans is considered ethically
unacceptable, and an artificial challenge cannot provide the valuable
information obtained by exposure to natural infection, which is modulated by
components of sand fly saliva. On the other hand, the large number of
participants needed for a field trial, due to the low expected vaccine efficacy
can be reduced only by the use of
vaccines with higher efficacies
(UNDP/World Bank/WHO Special Programme), such as second-generation ones formulated with potent adjuvants.
Recently,
it has been reported that photodynamic vaccination of hamsters with the
suicidal-mutants reduced the parasite loads by 99% and suppressed the
development of disease significantly for up to 180 days. These suppressions
were accompanied by an increase in delayed-type hypersensitivity response and Leishmania-specific lymphoproliferation
as well as in the levels of splenic iNOS, IFN-g and IL-12 expressions and of Leishmania-specific IgG2 in the serum.
Vaccines using recombinant viruses and bacteria as delivery
vehicles
Another
approach to second-generation vaccines is to use live recombinant bacteria or
virus expressing Leishmania parasite
antigen; the bacteria and virus serve as expression carrier and adjuvant
system. These vaccines have limited practical application. Examples of bacteria
vaccines are: L. major GP63 surface protease, a major Leishmania antigen, cloned in Salmonella
thypymurium mutant or in BCG; the LCR1 L.
chagasi antigen (similar to a T.
cruzi flagellar protein) in BCG and the KMP-11 (kinetoplastid) antigen in
attenuated tachyzoites of Toxoplasma
gondii. Examples of vaccines based on virus are Vaccinia virus expressing
the G46/M-2/PSA-2 promastigote surface protein which protects against L. amazonensis; or Vaccinia expressing
the L. infantum LACK antigen
(parasite analogue to the receptor for activated mammalian kinase C) which in
prime boost vaccination, protects mice against L. major and dogs against L. infantum infection.
6. Vaccines Based on
Purified Leishmania Antigens
A
further approach to second-generation vaccines includes the purified Leishmania sub-fractions. Integral
membrane proteins and non-membranous soluble proteins of L. donovani showed immunostimulatory cellular responses in cured Leishmania-infected patients and
hamsters. Our group have reported earlier that the soluble proteins from
promastigote of L. donovani ranging from 68 to 97.4 kDa (F2-fraction), induced
Th1-type cellular responses in cured Leishmania
patients and hamsters with significant prophylactic efficacy. Induction of
Th1-type cellular responses was also observed in cured/exposed Leishmania-infected patients and
hamsters against poly proteins of soluble L.
donovani promastigotes ranging from 89.9-97.1 kDa.
Several
other proteins or lipophosphoglycan have been used to assess their
immunogenicity, but because of difficulties in their mass production, they
never advanced to Phase II or Phase III trials. In the first Phase III trial
with a second-generation dog vaccine, Dunan et
al. using a semi-purified lyophilized protein preparation from L. infantum (94-67 kDa), paradoxically
achieved a significantly higher rate of infection in the vaccinated group than in the control group. This
vaccine then, while effective in murine models, did not induce protection
against canine kala-azar in the field.
Two
dog vaccines achieved successful results in Phase III trials: the FML-saponin
and the LiESAp-MDP vaccines. The glycoproteic enriched preparation of L. donovani promastigotes, named FML
(Fucose-Mannose ligand), antigenic for human and dogs, was formulated with
Quillaja saponaria saponin and passed Phase I—III trials to became the
Leishmune licensed vaccine in
The
other second-generation vaccine, LiESAp, composed of the 54 kDa excreted
protein of L. infantum plus MDP,
protected dogs in a kennel assay against L.
infantum infection. Parasites were
detected in the bone marrow of 3/3 placebo treated controls, while they were
absent in 0/3 vaccinated dogs. A double-blind randomized trial was further
performed with LiESAp + MDP in naturally exposed dogs in
7. Recombinant Antigens
The last approach in second-generation vaccines is the use of
recombinant proteins that were intensively tested since the 1990s. The Leishmania
recombinant vaccine candidates were assayed either alone, or in
combination, or as polyproteins or chimeras. In order to develop protection,
most of them needed to be formulated with adjuvants, or delivered by bacteria.
LeIF and HASPB1 proteins are the
exceptions. While most recombinant proteins were assayed for their
immunogenicity and protective potential in mice models, only a few of them
advanced to monkey trials for CL, to kennel assays in dog model against VL or
to pre-clinical studies in humans. None has advanced to dog’s Phase III trials.
The recombinant proteins were used against all forms of the disease and all
parasite species. In contrast to the results for first and second-generation
vaccines with native antigens, the results of protection developed by each
recombinant formulation in mice do not allow determination of VE values, since
no exposure to natural infection occurred. In this review, we considered for
comparison of protection due to recombinant or DNA vaccines, the reported
reduction of LDU or of limited dilution values in vaccinated mice compared to
untreated controls. In the specific case of CL mouse models, the results were
obtained from each report as the percent of the size of the footpad lesion of
the vaccinated animals compared to that of the untreated saline control, at the
latest time point when data for the saline control was still available. The
mean average of parasite reduction for all the assays with recombinant antigens
is 68.02% (IC 95%, 58.32—77.71). An interesting approach is the induction of
protective immunity by a polyprotein vaccine formulation. The TSA
(thiol-specific antioxidant) and LmSTI1 (L. major stress inducible
protein 1) are protective for mice and monkeys against CL, although the use of
recombinant adjuvant IL-12 is not recommended at present because it may promote
immune disorders and fail to induce long-term immunity. The multicomponent
Leish-111f fusion protein containing the antigens TSA, LmSTI1 and LeIF (Leishmania
elongation initiation factor), in formulation with MPL-SE9 and squalene,
protect mice against CL and VL but, in combination with MPL-SE or AdjuPrime,
was only immunogenic in dogs challenged with L. chagasi and L. infantum (MML), and failed to
prevent Leishmania infantum natural infection, or disease progression in
dogs in an open kennel trial. These dogs
received two courses of three-dose vaccine with 1 year interval.
Nevertheless, Leish111f + MPL-SE showed to be safe, immunogenic, and
reactogenic in healthy volunteers in the US and in patients of CL and ML in
Brazil and in Peru, respectively, while vaccination with the Leish111f
components, Leishmania heat shock protein 83 (Lbhsp83) and GM-CSF,
combined to chemotherapy, led to clinical improvement and complete cure of six
human patients with MCL. Furthermore, the H1 histone that protected mice and monkeys against CL, the HASPB1
(hydrophilic acylated surface protein B1) or both in combination with
Montanide, and the protein Q, a chimeric antigen composed of the genetic fusion
of five fragments of the acidic ribosomal protein Lip2a, Lip2b, P0 and the
histone H2A used with BCG developed partial protection against CVL in dogs
against infection and at the clinical level. Finally, poloxamer 407 adjuvant +
CPb, but not CPa was able to protect mice against CL whereas both in combination with IL-12 and
QuilA, failed to protect dogs from L. infantum infection, probably due
to the low concentration of the adjuvants (50 ug in each vaccine dose). As
single candidate recombinant vaccines tested in mice, the L. major LACK
with IL-12 was less efficient than the plasmid DNA encoding LACK, the
recombinant PSA2 antigen (parasite surface antigen 2) in ISCOMs or with C.
parvum, induced a Th1 response but not protection against CL, while the
LCR1 protein and the amastigote-specific
A2 antigen conferred protection to VL. The nucleoside hydrolase of L.
donovani (NH36) is an essential enzyme that releases DNA bases from
imported nucleosides, allowing the parasite to construct its own DNA, since Leishmania
parasites lack the de novo purine biosynthesis pathway. NH36 is the
main antigen of the FML complex that
constitutes the Leishmune® vaccine for CVL. The recombinant NH36 in combination
with Quillaja saponaria Molina saponin protected mice against L.
chagasi and L. mexicana infection through a Th1 response. In 1996,
the WHO-TDR program organized comparative studies of several leading
recombinant proteins known at that time. In 2001, the fourth Meeting on
Second-Generation Leishmania vaccines evaluated these studies and
discussed trends in Leishmania vaccine development. While most antigens
induced lymphocyte proliferation and IFNg secretion in naıve mice and human patients, the
only antigens that protected mice against CL were the MIX, LACK, 4H6 and FPA.
No assays were performed against VL and no quantification of reduction of
parasitic load was done. The main detected problem was the lack of stability
and potency during the transportation of the antigens, suggesting potentially
severe problems with future scale-up of recombinant vaccine production.
8. Candidates
for Third-generation Vaccines:
Compared to recombinant protein vaccines, DNA vaccines are
much more stable and have the advantage of their low cost of production, no
need of cold chain for distribution and flexibility of combining multiple genes
in a simple construct. The mechanism by
which DNA vaccination generates potent immune responses appears to be through
the activation of innate immune responses by the non-methylated CpG sequences
of bacteria and to the intense replication within the host, leading to the
expression of the recombinant proteins for longer periods. Use of DNA vaccines raises both humoral and
cellular immunity (Fig.1).
The most-studied antigens were those previously assayed as
recombinant proteins . Most of them were
tested as single vaccines, and some, as combination of genes or as heterologous
prime-boost (HPB), which involves an injection of the DNA vaccine followed by
an injection of the recombinant protein or a Vaccinia virus expressing the
recombinant protein. Adjuvants were added to formulations in only two studies.
Protection was observed in vaccines using all the tested plasmids, , with the
exception of pMOK in a dog assay and of pcDNA3 in a mice assay. Most trials were performed in mice
against CL and VL, and some in the hamster against VL and in dogs against CVL.
Fig.1. Use of DNA vaccines raises both humoral and cellular immunity. The
injected gene is expressed in the injected muscle cell and in nearby APCs. The
peptides from the protein encoded by the DNA are expressed on the surface of
both cell types after processing as an endogenous antigen by the MHC class I
pathway. Cells that present the antigen in the context of class I MHC molecules
stimulate development of cytotoxic T cells. The protein encoded by the injected
DNA is also expressed as a soluble, secreted protein, which is taken up,
processed, and presented in the context of class II MHC molecules. This pathway
stimulates B-cell immunity and generates antibodies and B-cell memory against
the protein. [Adapted from Weiner and Kennedy (1999)].
The most striking characteristic of the DNA vaccine studies is that about a half
of them used for challenge non-virulent
(NV) Leishmania strains, grown in
liquid culture media and at a low number of parasites, while the rest used virulent [V] parasites isolated
from infected animals. This
heterogeneity of protocols determined a high variation of percents of reduction
of parasite load, with a mean average of 59.24% (IC 95% 47.75-70.73). The different codified antigens could be the
responsible for the different degrees of protection. However, controversial results
for the same antigen refute this idea.
Protection did not correlate to the type of expression vector (p >
0.05), or to the addition of IL-12. It seems to be easier to achieve protection
in isogenic mice than in dogs. Most of the mouse investigations used 100
μg of plasmid, while protection due to CP proteinase vaccine required 200
μg. These results suggest the need for higher plasmid concentrations to
achieve efficacy. DNA vaccines are
indeed protective, although to date no Phase III trial data are available. A
lot of interest was though generated in vaccine development against
leishmaniasis in recent years, with studies going on in the labs on
experimental models. The LACK, LeIF,
TSA, LmSTI1, H1, CpA + CpB, KMP11 and NH36
are the most promising candidates that may find a place in the forth coming years , since they have
already been tested in more animal models.
In mice, LACK DNA induced a Th1 response
that protected against infection by L. major, but not L. donovani. Even truncated portions of the
LACK gene and PSA2 gene were superior to GP63 and p20 against L. major infection, and immunization with
HPB-LACK protected mice against VL and dogs from
CVL. The immunization resulted in an increase in IFN-γ and IL-12
expression, lymphocyte proliferative response, IgG2 to IgG1 ratio while it led
to decreases in clinical symptoms, number of parasites in target tissues, and
IL-4 expression.
Vaccination with 200 μg of KMP11
protected hamsters against VL through a mixed cytokine Th1/Th2 response, while
a cocktail of plasmid DNA encoding KMPII, TRYP, LACK, and GP63 (200 μg of each plasmid) did not protect dogs against L. infantum virulent challenge.
In spite of the many genes identified as vaccine candidates,
the slow knowledge transfer from the laboratory to industry, the GMP
regulations that dampen down the industrial interest, the poorly developed
biotechnology industry and the lack of scientists in regulatory agencies of
underdeveloped countries, the ethical constraints on research in animals and
the increasing dog-chemotherapy in Europe, where human leishmaniasis is less
frequent, contribute to the continuous use of first-generation and live
vaccines, and to the delay in the arrival of combined DNA vaccines to Public
Health.
Based
on
1.
Handman, E., 2001.
Leishmaniasis: current status of vaccine development. Clin Microbiol Rev 14,
229-243.
2.
Kumari, S., Kumar, A.,
Samant, M., Sundar, S., Singh, N., Dube, A., 2008a. Proteomic approaches for
discovery of new targets for vaccine and therapeutics against visceral
leishmaniasis Proteomics - Clinical Application
3, 372-386.
3.
Palatnik-de-Sousa, C.B.,
2008. Vaccines for leishmaniasis in the fore coming 25 years. Vaccine 26,
1709-1724.
4.
Kumari, S., Samant, M.,
Khare, P., Sundar, S., Sinha, S., Dube, A., 2008c. Induction of Th1-type
cellular responses in cured/exposed Leishmania-infected patients and hamsters
against polyproteins of soluble Leishmania donovani promastigotes ranging from
89.9 to 97.1kDa. Vaccine 26, 4813-4818.
Antileishmanial Potential of Indian Medicinal Plants
Sheela Tandon
Documentation & Library Services,Central Drug Research Institute,
Leishmaniasis, a vector-borne parasitic disease resulting from
infection of macrophages by obligate intracellular parasite of genus leishmania. Generic pentavalent
antimonials have been the main stay for therapy in the endemic regions because
of its efficacy and cost effectiveness. However, the growing incidence of
resistance for the pentavalent antimony complex in endemic and non-endemic
regions has seriously hampered their use in these regions. The second line of
drugs, such as amphotericin B, paromomycin and miltefosine are the other
alternatives, but they merely fulfill the desired requirements of a safe drug.
The recent researches focused on plants have shown a wise way to get a true and
potentially rich source of drug candidates against leishmaniasis. In this article we have tried to
highlight the role of selected “Indian Plants” in the management of
leishmaniasis.
Selected Indian
Medicinal Plants with Anti-leishmanial activity
|
Plant Name |
Active Component |
Reference |
|
Acanthus illicifolius |
2-Benzoxazolinone |
Planta
Medica (1994), 60(2), 187-8 |
|
Annona muricata |
Ethylacetate
extract |
Fitoterpia,
(2000), 71(2), 183-6 |
|
Allium sativum |
Garlic
extract |
Scand.J.
Immunology, (2007), |
|
Aloe vera |
Leaf
exudate |
Parasitolology
Res, (2008) |
|
Alstonia macrophylla |
Aqueous
and methanolic extract |
J
Ethanopharmacology, (2003) 89(2-3), 185-91 |
|
Andrographis paniculata |
Xanthones |
Phytotherapy
Res., (2009), |
|
Artemisia annua |
Artemisinin |
J.
Med. Microbiol. (2007), 56(9), 1213-8 |
|
Asparagus racemosus |
Racemoside
A |
J.Med
Microbiol, (2007) Sept, 56 (pt 9), 1196-204 |
|
Azadirachta indica |
Methanol
extract |
Turkiye
Parazitol Derg, (2005) 29(1), 3-6. |
|
Bacopa monnieri |
Bacopasaponin
C |
Drug
Deilv., (2002), 9(1), 55-62 |
|
Berberis aristata |
Berberine |
Exp.
Parasitology, (1985), 60(3), 404-13 |
|
Careya arborea |
Arborenin Desacylescin
III |
Phytochemistry,
(2006), |
|
Cassia fistula |
Sterols |
Phytotherapy
Res., (2007) |
|
Chenopodium ambrosioides |
Essential
oil, Hydroalcoholic crude extract |
J,
Ethanopharmacology, (2008), |
|
Crotolaria ramosissima |
Chromeno
dihydrochalcones |
Biorg.
Med. Chem. Lett. (2004) Aug,2, 14,15, 3913-6 |
|
Curcuma longa |
Curcuminoids |
Arzneimittelforschung.,
(2002), 52(9), 695-8. |
|
Cympogon citratus |
Essential
oils |
Parasitological
Res.,(2008), Dec 16 |
|
Desmodium gangeticum |
Aminoglucosyl
glycerolipid, Cerebroside |
Bioorg.
Med. Chem. Lett., (2005), Oct. 15, 15(20), 4543-6. |
|
Dysoxylum binectariferum |
Chloroform
fraction |
Phytomedicine,
(2007), Jan14(1) 36-42. |
|
Glycyrrhiza glabra |
(E)-1-[2,4-dihydroxy-3-(3-methyl-3-butenyl)phenyl]-3-[4-hydroxy-3-(3-methyl-2-butenyl]phenyl-2-propene-1-one
(Chalcone) |
Planta
Med., (1994), |
|
Kalanchoe pinnata |
Aqueous
extract(Flavoinoid-quercitrin) |
International
Immunopharmacology, (2008) 8(12) 1616-1621. |
|
Myristica malabarica |
Malabaricones |
Phytotherapy
Res, (2007), |
|
Nyctanthus arbor-tristis (Night Jasmine) |
Calceolarioside
A |
Planta
Medica, (2008),74(5) 503-8 |
|
Ocimum gratissimum |
Essential
oil |
Parasitol.
Res., (2008) |
|
Picrorhiza kurrcoa |
Picroliv
|
Life
Sci., (1998), 63(20), 1823-34. Acta
Trop. (2005), 94(1), 41-7 |
|
Piper betle |
Ethanolic
extract |
Parasitological
Res. (2008), |
|
Swertia chirata |
Amarogentin,seco-irridoid
glycoside |
J.Antimicrobial
Chemotheropy, (1999) |
|
Tephrosia pumila |
Pumilanol(isoflvan-4-ol) |
Phytochemistry
Letters, (2008) 1(4), 175-178. |
|
Tinospora sinensis |
Ethanolic
extract,butanol fraction |
Parasitol
Res. (2008), |
The drugs used currently for the treatment of Kala-azar cause
severe toxic side effects and acute immunosuppression in the treated
individuals. Picroliv is a standardized mixture of irridoid glycosides prepared
from the alcoholic extract of the root and rhizome of Picrorhiza kurroa has shown strong hepatoprotective activity
against several models of hepatotoxicity. Immuno-stimulant activity of Picrorhiza kurroa and its protective
action against Leishmania donovani
infection in hamsters is well documented.
A study was undertaken to evaluate the effects of Picroliv
(12.5 mg/kg x 7 days oral) and in combination with sodium stibogluconate (SSG)
on parasitemia, lipid peroxidation and hepatic marker enzymes of golden
hamsters during Leishmania donovani
infection. The results indicated a marked hepatopective effect of Picroliv in
terms of biochemical markers and a significant antileishmanial activity
implying that it can be utilized as an adjunct to chemotherapy or in
combination therapy of Kala azar along with sodium stibogluconate.
In further studies, Picroliv in combination with miltefosine,
showed promising results as an orally effective antileishmanial drug against
experimental Visceral leishmaniasis(VL). Miltefosine,
an alkyl phospholipids compound, is the first orally effective drug, which has
shown 98% cure rate of VL patients during phase III clinical trial in
The leaves of Piper betle (locally known as Paan) have long
been in use in the Indian indigenous system of medicine for its antimicrobial
properties. In a study ethanolic extract of leaves of Piper betle exhibited
antileishmanial activity in both promastigotes and amastigotes with IC50 values
of 9.8 and 5.45 microg/ml, respectively importantly, it was accompanied by a
safety index of ›12-fold. This leishmanicidal activity of PB was mediated via
apoptosis as evidenced by morphological changes, loss of mitochondrial membrane
potential, in situ labeling of DNA fragments by terminal deoxyribonucleotidyl-transferase-mediated
deoxyuridine triphos-phate nick and labeling, and cell cycle arrest at the
sub-G0/G1 phase.
Aloe vera has wide
spread use in health products. Various studies on Aloe vera have shown its effectiveness in four Leishmania donovani strains in both promastigotes and amastigotes.
In amastigotes the killing by Aloe vera was
facilitated through its induction of nitric oxide in leishmania infected
macrophages. The safety index was good as AVL upto 300 microg/ml remained
non-toxic to monocytes and macrophages.
It has been shown that garlic (Allium sativum) extract modulates immune responses. Control of Leishmania is associated with a Th1-type
immune response and garlic extract has been reported as a Th1 immunomodulator
in BALB/c mice infected with Leishmania
major. In a study, the effect of garlic extracts on L. mexicana infection in vivo
and in vitro. Garlic extract reduced
footpad lesions in L. mexicana-infected
BALB/c mice by inducing IFN-gamma production from T cells. In vitro, garlic extract reduced macrophage infection through
induction of nitric oxide (NO) production. Garlic extract may thus act on both
T cells and macrophages to stimulate IFN-gamma production and NO synthesis for
parasite killing. A 10- to 14-kDa fraction was identified as responsible for
the in vitro effect of the whole
extracted and may lead to the identification of novel immunomodulating drugs
and therapeutic alternatives for the treatment of leishmaniasis.
The treatment of leishmania is difficult because of the
intramacrophagic location of the infectious form. Victims of this illness
present an immune deficiency and are not able to eliminate the parasites
through a natural mechanism of defense. In the absence of vaccine there is an
urgent need for effective drugs to replace/supplement those in current use. The
plant kingdom is undoubtedly valuable as a source of new, safe and more
effective medicinal agents.
Leishmaniasis:
A Neglected Tropical Disease
Sheela Tandon & VK Vohra
Documentation
& Library Services, Central Drug Research Institute,
The neglected tropical diseases are a group of 13 major
disabling conditions that are among the most common chronic infections in low
income populations of
The 13 parasitic and bacterial infections known as the NTDs
include three soil-transmitted helminth infections (ascariasis, hookworm
infection, and trichuriasis), lymphatic filariasis, onchocerciasis,
dracunculiasis, schistosomiasis, Chagas’ disease, human African
trypanosomiasis, leishmaniasis, Buruli ulcer, leprosy, and trachoma. An
expanded list could include dengue fever, the treponematoses, leptospirosis,
strongyloidiasis, foodborne trematodiases, neurocysticercosis, and scabies, as
well as other tropical infections. The parasitic and bacterial diseases identified
as being neglected are among some of the most common infections in the
estimated 2.7 billion people. The Global Network for Neglected Tropical
Diseases – the first ever global effort to combat NTDs.
Table 1: The Major Neglected Tropical Diseases Ranked by Prevalence
|
Disease |
Global Prevalence
(millions) |
Population at Risk |
Regions of Highest
Prevalence |
|
Ascariasis |
807 |
4.2
billion |
|
|
Trichuriasis |
604 |
3.2
billion |
Sub-Saharan
|
|
Hookworm
infection |
576 |
3.2
billion |
Sub-Saharan
|
|
Schistosomiasis |
207 |
779
million |
Sub-Saharan
|
|
Lymphatic
filariasis |
120 |
1.3
billion |
|
|
Trachoma |
84 |
590
million |
Sub-Saharan
|
|
Onchocerciasis |
37 |
90
million |
Sub-Saharan
|
|
Leishmaniasis |
12 |
350
million |
|
|
Chagas’
disease |
8-9 |
25
million |
|
|
Leprosy |
0.4 |
ND |
|
|
Human
African trypanosomiasis |
0.3 |
60
million |
Sub-Saharan
|
|
Dracunculiasis |
0.01 |
ND |
Sub-Saharan
|
|
Buruli
ulcer |
ND |
ND |
Sub-Saharan
|
*ND
denotes not determined. (Source: N. Engl. J. Med., 357, 10;
1. Life Cycle
The parasitic protozoa of genus, Leishmania arethe pathogenic agents responsible for leishmaniasis.
During their complex life cycle, Leishmania
parasites are exposed to different extra- and intracellular environments. These
organisms are digenetic parasites with two basic life cycle stages: one
extracellular stage within an invertebrate host (Phlebotomine sand fly) and one
intracellular stage within a vertebrate host. Thus, the parasite exists in two
main morphological forms, amastigotes and promastigotes, which are found in
vertebrate hosts and invertebrate hosts, respectively.
1a.Stages in
the Invertebrate Host
The invertebrate hosts or vectors are small insects of the
order Diptera, belonging to the
subfamily Phlebotominae. They are
commonly called phlebotomine sand flies. Of the six genera described, only two
are of medical importance: Phlebotomus
of the ‘Old world’, divided into 12 subgenera, and Lutzomyia of the ‘
Within the intermediate host, Leishmania develops as promastigote forms, elongated motile
extracellular stages possessing a prominent free flagellum. Nevertheless, a
variety of different promastigote forms have been distinguished on
morphological grounds.
1b.Stages in
the Verebrate Host
Proliferative promastigotes differentiate into infective
metacyclic promastigotes in the fore guts of sand fly.Parasites are inoculated
by the vector as flagellate promastigotes enter the mammalian host, where they
infect macrophages, differentiating into nonmotile amastigotes and multiplying
as such. One of the most remarkable accomplishments of Leishmania is that they successfully parasitize the mammalian cells
that are responsible for killing invaders: the macrophages. Leishmania are extremely successful
parasites and natural infections are found in many different orders of mammals-rodents,
canids, edentates, marsupials, procyonids, primitive ungulates and primates.
All these mammals are considered as potential reservoirs of the disease. Humans
are possible hosts of these parasites, but in the majority of cases they are
considered to be accidental hosts.
In the vertebrate host, the parasite evolves into an
amastigote form. Amastigotes are ovoid (2.5-5 µm diameter), nonmotile
intracellular stages.They do not have a free flagellum and are located in the
parasitophorous vacuoles of the host’s macrophages.
The life cycle starts when a parasitized female sand fly takes
a blood meal from a vertebrate host. As the sand fly feeds, infective
promastigote forms (metacyclic promastigotes) enter the vertebrate host via the
insect’s proboscis. The promastigotes are then phagocytosed by macrophages in
which they metamorphose into amastigote forms and reproduce by binary fission.
They increase in number until the cell eventually bursts and then infect other
phagocytic cells to continue the cycle.
2.Taxonomy
A modern scheme of classification of Leishmania is shown in Figure 1.
The distribution area of the leishmaniases has been broadly
subdivided into the ‘
3. Clinical Expressions in Humans
When humans are bitten by an infective
sand fly, parasite
inoculation can lead to the development of leishmaniasis but can also have no
effect on health. The rate of asymptomatic carriers (infected individuals
without clinical manifestations) is not accurately known, but different studies
have suggested that it may be higher than expected.
. In humans, the disease occurs in at least four major forms:
cutaneous, diffuse cutaneous, mucocutaneous, and visceral.
Cutaneous
leishmaniasis is caused by various
species of leishmania and a wide variety of clinical presentations is possible.
A classical lesions starts as a nodule at the site of inoculation. A crust
develops centerally which may fall away exposing an ulcer which heals
gradually. The disease ranges from single, self-healing lesions which are
troublesome but not a threat to life, to multiple, deep and distructive ulcers
which cause considerable disfigurement and disability. Its most severe form, recidivans leishmaniasis, is very
difficult to treat, long-lasting, destructive and disfiguring.
Diffuse cutaneous
leishmaniasis occurs in individuals with a defective cell mediated immune
response. It occurs occasionally following infections with L. aethiopica or members of the L.
mexicana complex and involves dissemination of the disease from the
original site of infection to distant skin sites. The lesions resemble
lepromatous leprosy do not heal spontaneously and are resistant to treatment. Because
of the devastating consequence to the patient, it is recognized as a special
public health problem.
Mucocutaneous
leishmaniasis, also known as espundia, causes extensive destruction of
oro-nasal and pharyngeal cavities with hideously disfiguring lesions,
mutilation of the face and great life-long distress for the patient. Ulceration
and erosion progressively destroy the soft tissue and cartilage of the
oronasal/pharyngeal cavity. Mutilation is severe and secondary bacterial
infections is frequent and can be fatal.
Visceral leishmaniasis,
also known as kala-azar, is the most severe form (nearly always fatal if left
untreated), characterized by undulating fever, loss of weight, splenomegaly,
hepatomegaly and/or lymphadenopathies and anaemia. It causes large-scale
epidemics with a high fatality rate. After recovery, patients may develop a
chronic
![]()
Subkingdom Protosoa
![]()
![]()
Order Kinetoplastida
![]()
![]()
Family Trypanosomatidae
Genus Crithida Leptomonas
Herpetomonas Blastocrithidia Leishmania
Sauroleishmania Trypanosoma Phytomonas
Endotrypanum
![]()
Subgenus Leishmania
Viannia
![]()
![]() Complex L.donovani L.tropica
L.major L.aethiopica L.maxicana
L.braziliensis L.guyanensis L.naiffi
L.lainsoni
Complex L.donovani L.tropica
L.major L.aethiopica L.maxicana
L.braziliensis L.guyanensis L.naiffi
L.lainsoni
![]()
![]()
![]()
![]()
![]()
![]()
![]()
Species L.archibaldi L.killcki
L.major L.aethiopica L.amazonensis L.braziliensis L.panamensis L.naiffi
L.lainsoni
L.chagasi L.tropica L.garnhami L.peruviana L.guyanensis


L.infantum
L.mexicana Non
pathogenic
For humans L.shawi No final
L.pifanoi
L.venezuelensis
L.gerbilli L.colombiensis
L.arabica L.equatorensis
L.turanica
L.forattinii
L.arisitidesi
L.enrietti
L.deanei
L.hertigi
Figure 1.
Taxonomy of Leishmania
(Source: Advances in Parasitology ,(2007), Vol. 64,
p.7)
Table 2: Major Leishmania
Species that Cause Disease in Humans
|
Species |
Clinical Syndrome |
Geographic Distribution |
|
SUBGENUS LEISHMANIA L. donovani complex L.
donovani sensu stricto L.
infantum sensu stricto L.
chagasi L. mexicana complex L.
mexicana L.
amazonensis L. tropica L. major L. aethiopica |
VL
(PKDL, OWCL) VL
(OWCL) VL
(NWCL) NWCL
(DCL) NWCL
(ML, DCL, VL) OWCL
(VL) OWCL OWCL
(DCL, ML) |
Central
and Central
Asia, India, Pakistan, southwestern Asia, Middle East, Turkey, North Africa,
Sahel region of north-central Africa, Ethiopia Sudan, Kenya |
|
SUBGENUS
VIANNIA L. (V.) braziliensis L. (V.) guyanensis L. (V.) panamensis L. (V.) peruviana |
NWCL
(ML) NWCL
(ML) NWCL
(ML) NWCL |
Central
and |
Abbreviations: VL Visceral Leishmanisis;
PKDL Post kala-azar dermal Leishmanisis; OWCL Old World Cutaneous Leishmanisis;
NWCL New World Cutaneous Leishmanisis; DCL Diffuse Cutaneous Leishmanisis; ML
Mucosal Leishmanisis (Source:
cutaneous form called
post-kala-azar dermal leishmaniasis (PKDL), which usually requires long and
expensive treatment. PKDL is normally a sequel to kala-azar treatment, although
some cases have been reported with no history of kala-azar. Five to fifteen
percent of visceral leishmaniasis patients in
It appears that the different clinical forms are closely
related to the adaptive immune response of the host, especially the euilibrium
between cellular and humoral immunity. The nature of the pathogen, notably the
species, seems to be a strong factor as well.
4. The Immune Response in Human Leishmaniasis
Since many individuals remain asymtomatic, it is obvious that
the natural immune response of humans can eliminate or control the parasites. Macrophages
are the first host cells to contact and be parasitized by Leishmania. They are key cells in the host immune defence. These
cells, as well as dendritic cells, present the parasite antigens to T cell
receptors, via the major histocompatibility complex (MHC) molecules. This is
the acquired immune response. This step is fundamental since it may influence
the type of immune response depending on the cytokine context and on the Leishmania peptides presented. Thus, Leishmania parasites have evolved
mechanisms to evade or interfere with antigen presentation processes, making it
possible to partially resist the T cell mediated immune responses.
These escape strategies appear complex and various since, in
humans, different patterns of immunological response are observed according to
the clinical manifestation and exposure to different Leishmania species. Briefly, different T cell responses are
observed among the different cutaneous forms of leishmaniasis: an absence of a
Th1 response (rather than presence of Th2) in diffuse cutaneous leishmaniasis;
a Th1 response in patients with self-healing lesions and a mixed Th1/Th2
response with high interferon-γ (IFN-γ) levels in patients with
mucocutaneous leishmaniasis. In visceral leishmaniasis, a mixed Th1/Th2
response is observed with production of IFN-γ along with interleukin-10
(IL-10). However, individuals with asymptomatic or subclinical infections of
visceralizing species of Leishmania
show peripheral blood mononuclear cell (PBMC) proliferation and production of
IL-2, IFN-γ and IL-12; in cured patients, both Th1 and Th2 clones
producing IFN-γ and IL-4 have been isolated.
5. Diagnosis: Diagnosis of leishmaniasis requires demonstration of the parasite. To
identifiy amastigotes by light-microscopic examination, the specimen obtained
from an infected site (e.g. thin smear, histologic section) should be stained
with Giemsa or another Romanovsky stain and presumptive amastigotes (
The Leishmania species that infect humans
are morphologically similar. They can be distinguished by isoenzyme analysis of
cultured promastigotes, determination of monoclonal antibody specificity, or
various molecular methods.
Indirect
immunologic methods for diagnosis include serologic assays and tests for Leishmania-specific cell-mediated
immunity (e.g. skin testing for delayed-type hypersensitivity reactions).
Traditional serologic assays (e.g. indirect fluorescent antibody testing) do
not reliably distinguish past from current infection, and no leishmanin skin
test preparation has been approved for use in the
6. Therapeutic
Options: For more than half a
century, the pentavalent antimonial (Sbv) compounds sodium
stibogluconate and meglumine antimonite have been the mainstays of
antileishmanial therapy.
The
traditional parenteral alternatives to Sbv – amphotericin B and
pentamidine isethionate – are generally considered more apt to induce serious
or irreversible toxicity. However, these agents, especially amphotericin B, are
being advocated for use in some situations, in part because of the benefits of
new formulations (e.g. lipid formulations of amphotericin B) and the decreasing
effectiveness of Sbv in some settings. With the apparent exception
of miltefosine, the other oral agents evaluated to date typically have at best
modest activity against some of the Leishmania
species. (Table 3)
7.
Prevention and Control: The
transmission of Leishmania species
typically is focal, in part
because of the limited flight range of sandflies;
these insects usually remain within a few hundred meters of their breeding
site. They
Table-3:
Parenteral and Oral Drugs for Treatment of Leishmaniasis
(Source:
|
Clinical Syndrome, Drug |
Route of Adminis-tration |
|
Visceral Leishmaniasis |
|
|
First-line therapy Pentavalent antimony Amphotericin B, lipid Formulation Alternatives Amphotericin B (deoxycholate) Paromomycin sulfate Pentamidine isethionate Miltefosine |
IV, IM IV IV IV, IM IV, IM |
|
Cutaneous Leishmaniasis |
|
|
First-line therapy Pentavalent antimony Parenteral
alternatives Pentamidine isethionate Amphotericin B (deoxycholate) Oral alternatives Fluconazole Ketoconazole Itraconazole Dapsone |
IV, IM IV, IM IV |
|
Mucosal Leishmaniasis |
|
|
First-line therapy Pentavalent antimony Amphotericin B (deoxycholate) Alternative Pentamidine isethionate |
IV, IM IV IV, IM |
rest in dark, moist places in habitats ranging from
deserts to rain forets; peridomestic sandflies rest in debris or rubble near
buildings. Vector control may be useful in some settings.
Personal
protective measures include avoiding outdoor activities when sandflies are most
active (dusk to dawn); using mechanical barriers such as screens and bed-nets
that keep out sandflies, which typically are about one-third the size of
mosquitoes; wearing protective clothing; and applying insect repellent to
exposed skin. Vaccine strategies are being investigated. Treating human cases
is an effective control measure only where humans are the primary reservoir
hosts of infectdion (e.g. of L. donovani
infection in
Based on
1.
Anne-Laure
Bahuls, Mallorie Hide and Franck Prugnolle Advances
in Parasitology, (2007), 64, p.1-109.
2. Peter J. Hotez, et al., The
Journal of Medicine, (2007), 357(10), 1018.
3.
16th
Edition,
Subscription
Form
To,
The Scientist-in-Charge
Documentation and Library
Services Division
CENTRAL DRUG RESEARCH
INSTITUTE
Post Box No. 173,
LUCKNOW-226 001,
Dear Sir,
I/My organization want to
be the annual subscriber of:
1. Drugs and Pharmaceuticals - Industry Highlights
2. Drugs and Pharmaceuticals - Current
R&D Highlights
I/We
am/are remitting the subscription amount of Rs./$ __________________ by Demand
Draft No. __________________ dated _______________ towards subscription of the
journal(s) as (1) Student/ Professional, (2) Educational/R&D Institution,
(3) Corporate Sector subscriber.
Name :
____________________________________________________
Address :
___________________________________________________
:
____________________________________________________
SIGNATURE WITH SEAL
Please send your payment by Demand Draft only in favour of
THE DIRECTOR,
CENTRAL DRUG RESEARCH INSTITUTE,
NEWS VIEWS
G8 Urged to Address Neglected Diseases
The World Health Organization and the independent Drugs for
Neglected Diseases initiative (DNDi) are urging the leaders of the world's
richest economies to address issues related to neglected tropical diseases as
part of the upcoming G8 summit in
A joint petition from the two groups says that improved
control programmes and funding for research into drug treatments and
diagnostics are badly needed for a variety .of chronically endemic diseases.
While international attention is now focused on HIV/AIDS,
malaria and tuberculosis, many other diseases place a large burden on poor populations and “are .still very
much neglected by the global public health agenda”, they say.
While the WHO is not part of the non-profit DNDi, its special
programme for research and training in tropical diseases (run together with the
United Nations' children's fund UNICEF and the World Bank) is a permanent
observer to the initiative.
Noting
that most infectious and parasitic diseases are actually preventable or
treatable, the petition highlights outstanding problems such as delivery
infrastructure and access to treatments. While some existing drugs are low-cost
and effective, in general simpler and safer treatments are needed, and there
are now no effective diagnostics and therapies for diseases such as
trypanosomiasis and leishmaniasis, the petition states."
(Scrip, 4.7.2008, p. 24)
Government
Plans Grants to Private Firms to Cure Neglected Diseases
The
government may soon give non-repayable grant-in-aid to pharmaceutical companies
for inventing new drugs for neglected tropical diseases like tuberculosis,
kala-azar, leprosy, malaria and filariasis.
This
is in addition to the existing support given to research by private companies.
Presently, the government gives grants-in-aid to public-funded research
institutions and soft loans to companies. A sum of Rs 45 crore
has been set aside for this purpose. The Department of Science and Technology (DST)
has moved a Cabinet note in this regard for consideration of the CCEA.
“Companies
tend to focus more on lifestyle diseases like obesity, cardiovascular, diabetes
and hypertension because of better return as these diseases are prevalent in
well-to-do families. But there is a need for more innovation in neglected
tropical diseases like diarrhoea, and kala-azar, which are mostly prevalent
among the lower classes of people who are unable to afford medicine,” a DST
official said adding that strategic public investment by the government in the
field would be the right answer as such investment in drugs will primarily be
undertaken with the objective of providing drugs to the poor at low cost
without profit motive.
Companies
like Ranbaxy Laboratories, Cadila Health Care, Alembic, Cadila Pharma, Dr
Reddy’s Laboratories and Torrent Pharma have asked the government to provide
such assistance.
DST
already runs a programme called drugs & pharmaceutical research programme
(DPRP) to support industry-institutional collaborative R&D projects and
infrastructure development for drug-related discoveries. Under the programme, DST
provides soft loans to pharma companies for R&D projects of 70% of total
project cost at a simple interest rate of 3% a year. Public-funded research
institutes, however, get complete grant-in-aid for such projects.
Under the new project,
government would extend the grant-in-aid to private companies also. The aid
would, however, be restricted to meet the recurring expenditure to be incurred
by the industry towards drug development for neglected tropical diseases.
Companies would not be allowed to use the aid for capital building or spending
on existing manpower.
(The Economic Times, 7.3.2008)
GSK Enters New
Neglected Disease Collaboration
GlaxoSmithKline and the Drugs for Neglected Diseases
Initiative (DNDi) have entered into a collaborative agreement to identify and
develop from existing GSK research programmes potential new treatments for
three major tropical diseases.
The tie-up, which will run initially for two years, will
target visceral leishmaniasis, human African trypanosomiasis (sleeping sickness)
and Chagas disease. The work will be done mainly at GSK's research facility in
Tres Cantos,
The hope
is to develop treatments with improved effectiveness, shorter and more
convenient administration regimens, fewer side-effects and lower costs,
overcoming some of the shortcomings of existing therapies, which are limited in
number and in some cases are facing resistance problems.
(Scrip, 14.3.2008, p. 4)
Gates Foundation Gives DNDi $26 Million for Neglected
Diseases
The Bill & Melinda Gates Foundation has given the
Geneva-based not-for-profit product development partnership, Drugs for
Neglected Diseases initiative (DNDi), 625.7 million overtime years to find new
medicines to treat human African trypanosomiasis (sleeping sickness) and
visceral leishmaniasis.
The DNDi already has several products in late-stage clinical
trials for these diseases, but they have a number of drawbacks, including
difficult administration routes for developing countries (intravenous or
intramuscular injections), toxicity and cost.
The grant from the Gates Foundation will enable DNDi to expand
its preclinical and lead optimisation drug discovery projects to develop new
and better drug candidates for human African trypanosomiasis and visceral
leishmaniasis, according to DNDi spokesperson Ann-Marie Sevcsik. DNDi plans to
select one lead candidate for each disease to take into Phase I clinical
trials. Sleeping sickness, a fatal disease if left untreated, threatens more
than 50 million people in 36 countries in sub-Saharan Africa, while visceral
leishmaniasis threatens 200 million in 62 countries.
61% of DNDi's money comes from
public/government donors, 33% from Medicines sans Frontiers, and the remainder
from other foundations and donors. To date, DNDi has secured €74 million from
donors (including the Gates' money), but says it needs a further €200 million
to deliver six to eight new treatments by 2014.
(Scrip No. 3321, 19.12.2007, p. 16)
R&D HIGHLIGHTS
Visceral leishmaniasis: advances in
treatment.
Maltezou
HC.
Recent Patents Anti-Infect Drug Disc. 3(3):192-8 (Nov., 2008)
Visceral leishmaniasis continues to be an important public
health problem worldwide. This vector-borne infection affects approximately
500,000 people annually with more than 50,000 associated deaths, a number that
among parasite diseases is surpassed by malaria only. Leishmaniasis was
recently selected by the World Health Organization for elimination by 2015.
Major obstacles for achieving this goal
include lack of an antileishmanial vaccine, wide-spread resistance to
pentavalent antimonials in the State of Bihar, India where half of cases
globally occur, and drawbacks of alternative antileishmanial drugs, including
prolonged administration, serious adverse effects, and high costs in poor
endemic areas.During the past decade, significant progress has been made
towards the development of new and less toxic antileishmanial agents, including
the oral agent miltefosine. Currently, there are several agents with
antileishmanial activity under investigation as well as patents that may
deserve further testing within combination regimens. In order to preserve the
activity of available antileishmanial agents, monitoring of their delivery,
response, and resistance should be implemented globally. Combination regimens
should be further investigated in large trials. The costs of antileishmanial
agents should be minimized in poor endemic areas where there are needed most.
Plasma sCD26 and sCD30 levels in cutaneous
leishmaniasis.
Shakib RJ et al.
Acta Trop. 2008 Oct 14.
CD26 and CD30 are surface molecules expressed on activated Th1
and Th2 cells, respectively. It is, therefore assumed that plasma levels of
CD26 and CD30 (sCD26 and sCD30) correlate with Th1 and Th2 response,
respectively. In this study, plasma levels of sCD26 and sCD30 in patients with
non-healing form of cutaneous leishmaniasis (CL) were measured and compared
with the levels of sCD26 and sCD30 in patients with healing form of CL and
healthy control volunteers. The results indicate that the plasma levels of
sCD26 and sCD30 are significantly (p<0.05) higher in patients with
non-healing form of CL than patients with healing form of CL or healthy
control. No significant difference is seen in the levels of sCD26 and sCD30 in
patients with healing form of CL in comparison with healthy control group. It
is concluded that sCD30 might be used as an indicator of a Th2 response in
patients with non-healing form of CL.
Amino acid residues of leishmania donovani cyclophilin key to interaction with its
adenosine kinase: biological implications.
Sen, B. et al.
Biochemistry,46(26),
7832 (
Cyclophilins (CyPs), by interacting with a variety of
proteins, often modulate their biological activities and thus have been
implicated in several cellular functions. However, mechanisms that determine
such interactions are poorly understood. Authors have earlier reported that an
endoplasmic reticulum (ER)-located cyclophilin (LdCyP) from the purine
auxotrophic parasitic protozoan Leishmania
donovani reactivated its adenosine kinase (AdK). The AdK-reactivating
property of LdCyP was however abolished at high ionic strength but not by
nonionic detergents. Modeling of LdCyP, based on its crystal structure solved
at 1.97 Å resolution, revealed several solvent-exposed hydrophobic and charged
residues. Mutagenesis of several of such solvent-exposed residues was performed
and their corresponding activities with regard to their (i) AdK reactivation
property, (ii) ability to form complex with the enzyme, (iii) capacity to
induce red shift in the intrinsic tryptophan fluorescence maxima of AdK, and
(iv) efficiency to withdraw the ADP inhibition from the AdK-mediated reaction
were compared to the wild-type protein. Results indicated that while the
replacement of R147 with either A or D severely impaired all of the above
characteristics displayed by the wild-type LdCyP, the effect of mutating K114
and K153 was although relatively less but nevertheless noticeable. Alteration of
other exposed hydrophobic and charged residues apparently did not have any
discernible effect. Under the condition of cellular stress, the ER-located
LdCyP is released into the cytoplasm with concomitant increase both in the
specific activity of the cytosol-resident AdK and the uptake of radiolabeled
Catalysis and structural properties of leishmania infantum glyoxalase ii:
trypanothione specificity and phylogeny.
SousaSilva,
M. et al.
Biochemistry, 47(1),
195 (
The glyoxalase pathway catalyzes the formation of D-lactate
from methylglyoxal, a toxic byproduct of glycolysis. In trypanosomatids,
trypanothione replaces glutathione in this pathway, making it a potential drug
target, since its selective inhibition might increase methylglyoxal
concentration in the parasites. Two glyoxalase II structures were solved. One
with a bound spermidine molecule (1.8 Å) and the other with D-lactate at the
active site (1.9 Å). The second structure was obtained by crystal soaking with
the enzyme substrate (S)-D-lactoyltrypanothione. The overall structure of
Leishmania infantum glyoxalase II is very similar to its human counterpart,
with important differences at the substrate binding site. The crystal structure
of L. infantum glyoxalase II is the first structure of this enzyme from
trypanosomatids. The differential specificity of glyoxalase II toward
glutathione and trypanothione moieties was revealed by differential substrate
binding. Evolutionary analysis shows that trypanosomatid glyoxalases II
diverged early from eukaryotic enzymes, being unrelated to prokaryotic
proteins.
Probing the structures of leishmanial
farnesyl pyrophosphate synthases: homology modeling and docking studies.
Mukherjee,
Prasenjit et al.
Journal of Chemical Information and
Modeling, 48(5), 1026 (
Leishmania donovani and
Leishmania major farnesyl
pyrophosphate synthase (LdFPPS and LmFPPS) are potential targets for the
development of antileishmanial therapy. The protein sequence for LdFPPS was
recently elucidated. Highly refined homology models were generated using the
protein sequences of LdFPPS and the closely related LmFPPS enzyme. A
ligand-refined model of LmFPPS with a bound bisphosphonate ligand was generated
using restraint-guided molecular mechanics followed by quantum
mechanics/molecular mechanics refinement. The ligand-refined model of LmFPPS
was further validated through extensive pose validation, enrichment, and other
docking studies involving known bisphosphonate inhibitors. The model was able
to explain the critical binding site interactions and site-directed mutagenesis
data obtained from experimental studies on related FPPS enzymes. The ligand-refined
model in conjunction with the validated docking protocol could be utilized in
the future for structure-based virtual screening and rational drug design
studies against these targets
Auricular leishmaniasis mimicking squamous
cell carcinoma.
J Laryngol Otol. 2008 Oct 28:1-4. [Epub ahead of print]
The authors report a rare case of auricular involvement by
leishmaniasis, in order to demonstrate the importance of thorough investigation
of cutaneous head and neck lesions, and also the importance of inclusion of
infections such as leishmaniasis in the differential diagnosis of auricular
lesions, especially in endemic areas.
A 42-year-old man with multiple lesions on his head, neck and
hands had the following lesions: a painful, crusted, 8 x 8 cm plaque with
indurated margins on the left parotid region and auricle; a red papule on the
right temporal region; an ulcerative lesion on the skin overlying the proximal
interphalangeal joint of the fifth finger of the right hand; and a bluish
papule on the neck. Although histopathological examination of the
Geimsa-stained specimen was misleading, a direct smear prepared from biopsies
showed amastigotes, and therapy resulted in complete recovery. Leishmaniasis
can be both under- or over-diagnosed.
Especially in endemic areas, parasitic causes of chronic infections
should always be kept in mind.
Multilocus microsatellite typing (MLMT)
reveals genetic homogeneity of Leishmania
donovani strains in the Indian subcontinent.
Alam MZ et al..
Infect Genet Evol. 2008 Oct 8. [Epub ahead of print]
In this population genetic study of Leishmania donovani parasites in the Indian subcontinent, 132
isolates obtained from patients in Bangladesh, India, Nepal and Sri Lanka
suffering from Kala-azar (100), post-Kala-azar dermal leishmaniasis (PKDL) (25)
and cutaneous leishmaniasis (CL) (2), and from 5 patients whose clinical
patterns were not defined, were analysed by using 15 hyper-variable
microsatellite loci. Multilocus microsatellite typing (MLMT) data were analysed
by using a Bayesian model-based clustering algorithm and constructing
phylogenic tree based on genetic
distances. In total, 125 strains from
Complexities of assessing the disease
burden attributable to leishmaniasis.
PLoS Negl Trop Dis. 2008;2(10):e313.
Epub 2008 Oct 29.
Among parasitic diseases, morbidity and mortality caused by
leishmaniasis are surpassed only by malaria and lymphatic filariasis. However,
estimation of the leishmaniasis disease burden is challenging, due to clinical
and epidemiological diversity, marked
geographic clustering, and lack of reliable data on incidence, duration, and
impact of the various disease syndromes. Non-health effects such as
impoverishment, disfigurement, and stigma add to the burden, and introduce
further complexities. Leishmaniasis occurs globally, but has disproportionate
impact in the Horn of Africa,
Characterizing the effect of pentamidine isothionate
on the immune system using mouse splenocytes as an experimental model.
Plaza DF et al.
J Immunotoxicol. 2007 Oct;4(4):279-85.
Evaluating immunomodulating effects is currently a fundamental
parameter when designing pharmaceutical products. Rather than wait for the
appearance of immunotoxic effects in patients, pre-clinical assays that
characterize these potential events in experimental models not only can
facilitate the understanding of how these phenomena arise, but sometimes reveal
findings that can lead to modifications in currently-accepted methodologies
used for estimating the pre-clinical assay predictive risk values. Pentamidine
isothionate, a drug used as a second option in treating leishmaniasis, has been
repeatedly shown in animals and humans to exert a toxicity at the renal,
cardiac and hepatic level; however, its immunotoxic effect have not been as
fully evaluated. The potential immunomodulatory effects of pentamidine on
splenocytes from two species of mice (BALB/c and ICR) were evaluated ex vivo in this study. The results here
indicated that there were significant differences in subpopulation profiles
between the strains even before treatment with the drug, with the variances
most apparent regarding the relative percentages of CD8(+) and CD19(+) cells in
splenocyte preparations. The data also showed that the drug preferentially
induced toxicity in BALB/c mice CD8(+)
and CD19(+) cells more so than in their ICR counterparts. Conversely, the ICR
cells seemed to be more susceptible to drug-induced spontaneous blastogenesis
than the cells from BALB/c hosts. The differential results seen here confirm
that: any potential immunomodulatory effect of a drug should be studied in at
least two different mouse strains during an evaluation of overall toxicologic
potential; and, there should be attempts to correlate phenotypical findings
with those of functionality before a drug can truly be deemed a safe compound.
Leishmania
major ascorbate peroxidase overexpression protects cells against reactive
oxygen species-mediated cardiolipin oxidation.
Dolai,
Subhankar et al.
Free
Radical Biology and Medicine, 45(11), 1520 (
Heme peroxidases are a class of multifunctional redox-active
proteins found in all organisms. Authors have recently cloned, expressed, and
characterized an ascorbate peroxidase from Leishmania
major (LmAPX) that was capable of detoxifying hydrogen peroxide.
Localization studies using green fluorescent protein fusions revealed that
LmAPX was localized within the mitochondria by its N-terminal signal sequence.
Subcellular fractionation analysis of the cell homogenate by the Percoll
density-gradient method and subsequent Western blot analysis with anti-LmAPX
antibody further confirmed the mitochondrial localization of mature LmAPX.
Submitochondrial fractionation analysis showed that the mature enzyme
(~3.6 kDa shorter than the theoretical value of the whole gene) was
present in the intermembrane space side of the inner membrane. Moreover,
expression of the LmAPX gene was increased by treatment with exogenous H2O2,
indicating that LmAPX was induced by oxidative stress. To investigate the
biological role of LmAPX we generated Leishmania cells overexpressing LmAPX in
the mitochondria. Flow-cytometric analysis, thin-layer chromatography, and IC50
measurements suggested that overexpression of LmAPX caused depletion of the
mitochondrial ROS burden and conferred a protection against mitochondrial cardiolipin
oxidation and increased tolerance to H2O2. These results suggest that the
single-copy LmAPX gene plays a protective role against oxidative damage.
Functional analysis and complex gene
rearrangements of the folate/biopterin transporter (FBT) gene family in the
protozoan parasite Leishmania.
Ouameur,
Amin Ahmed et al.
Molecular and Biochemical Parasitology, 162(2),
155 (Dec., 2008)
The protozoan parasite Leishmania is a folic acid auxotroph.
Previous work has led to the characterization of the main folate transporter
FT1. FT1 is part of the folate/biopterin transporter (FBT) family and
Leishmania with its 14 members is, of all sequenced organisms, the one with the
most FBTs. Authors developed a real-time TaqMan RT-PCR assay to follow the
expression of these FBT genes during growth phases, life cycles and in
methotrexate-resistant mutants of Leishmania
infantum. FT1 is expressed preferentially in the logarithmic phase which is
consistent with the higher accumulation of folate in that stage. FT1 RNA levels
even seemed to be related to folate concentration in the medium. Several of the
FBT genes were expressed preferentially in the stationary phase of growth, a
stage with minimal folate accumulation. It suggests that these FBT members may
transport other related substrates. Resistance to methotrexate is associated
with FT1 inactivation and upregulation of other FBT genes. Inactivation of FT1
is due either to a gene deletion mediated by homologous recombination between
conserved FBT sequences or by segmental gene conversion. This study highlighted
the multiplicity of FBT genes in Leishmania, their complex RNA expression, and
novel gene rearrangements associated with FT1 inactivation and antifolate
resistance.
Stimulus (polyphenol, IFN-[gamma],
LPS)-dependent nitric oxide production and antileishmanial effects in RAW 264.7
macrophages.
Kolodziej,
Herbert et al.
Phytochemistry, 69(18),
3103 (Dec., 2008)
The
effects of interferon (IFN-[gamma]), lipopolysaccharide (LPS), and some
polyphenols as individual stimuli, as well as in various combinations on NO
production in non-infected and infected macrophage-like RAW 264.7 cells were
investigated, with emphasis on the NO/parasite kill relationship. In
non-infected and in Leishmania parasitized cells, gallic acid significantly
inhibited the IFN-[gamma] and LPS-induced NO detected in the supernatant. This
effect was less prominent in IFN-[gamma]- than in LPS-stimulated cells.
Interestingly, and in contrast to non-infected cells, gallic acid inhibited NO
production only when added within 3 h after IFN-[gamma] + LPS.
Addition of gallic acid following prolonged incubation with
IFN-[gamma] + LPS periods (24 h) no longer inhibited, sometimes
even enhanced NO release. Notably, an excellent NO/parasite kill relationship
was evident from all the experiments. This study was extended to a series of
polyphenols (3-O-shikimic acid, its 3,5-digalloylated analogue, catechin, EGCG,
and a procyanidin hexamer) with proven immunostimulatory activities. Although
these compounds themselves were found to be weak NO-inducers, the viability of
intracellular Leishmania parasites was considerably reduced. Furthermore, their
dose-dependent effects on macrophage NO release was determined in the presence
of IFN-[gamma] and/or LPS. Again, non-infected and infected cells differed
significantly in the NO response, while inhibition of IFN-[gamma] and/or
LPS-induced NO production by the tested polyphenols strongly depended on the
given time of exposure and the sequence of immunological stimuli. A strong
inverse correlation between NO levels and intracellular survival rates of
Leishmania parasites supported the assumption that the observed inhibition of
NO was not simply due to interference with the Griess assay used for detection.
Anti-histone antibodies in dogs with
leishmaniasis and glomerulonephritis.
Ginel,
Pedro J. et al.
Research in Veterinary Science, 85(3),
510 (Dec., 2008)
The association between serum anti-histone antibodies and
glomerulonephritis was studied in 43 dogs with leishmaniasis (Leishmania infantum). Dogs with
increased serum creatinine levels and urine protein-creatinine ratio >1 were
considered to have glomerulonephritis. Moderately elevated anti-histone
antibodies were found in 38.89% (7/18) of infected dogs without glomerulonephritis,
whereas 88% of dogs with glomerulonephritis (22/25) showed moderate or strongly
elevated anti-histone antibodies. Prevalence of positive anti-histone
antibodies reactions and mean serum concentration was significantly higher
(P < 0.001; P < 0.0001) in infected dogs with
glomerulonephritis. Correlation between anti-histone antibodies and urine
protein-creatinine ratio was significant when groups were analysed together
(P < 0.046). Positive predictive value for glomerulonephritis of
positive anti-histone antibodies was 88%. In conclusion, high anti-histone
antibodies are significantly associated with glomerulonephritis. Although other
factors must be involved, dogs with moderate or strong positive anti-histone
antibodies reactions may have a higher probability to develop glomerular
lesions in canine leishmaniasis.
Altered peptide ligands can modify the Th2
T cell response to the immunodominant 161-175 peptide of LACK (Leishmania
homolog for the receptor of activated C kinase).
Jensen,
Molecular Immunology
Following Leishmania
major infection, the early LACK (Leishmania homolog of receptors for
activated C kinase)-induced IL-4 response appears to determine disease
susceptibility in BALB/c mice. Therefore, authors sought to manipulate the
pathogenic T cell responses to the immunodominant epitope with the use of
altered peptide ligands (APLs). Conservative and non-conservative substitutions
for each amino acid of the LACK 161-175 peptide determinant were tested for
their stimulatory capacity in four different LACK-reactive T cell systems. From
these results, authors propose a likely LACK 163-171/I-Ad core peptide register
and show that APLs with changes at putative T cell receptor (TCR) contacts
provide the greatest potential for immune deviation. In particular, the
TCR-contact H164V APL expanded Th1 cells upon in vitro recall of naïve
splenocytes from LACK-specific BV4 T cell receptor transgenic mice and
stimulated IFN-[gamma] secretion from a Th2-committed LACK-reactive T cell line.
Authors also observed that non-conservative substitutions flanking the core
determinant had strong agonistic effects for proliferation and Th1/Th2
modulation. However, upon immunization, the H164V APL considerably
downregulated proliferation and cytokine responses to the wild type LACK
161-175 peptide, while immunization with the weak agonist, MHC contact APL
S171K, increased the IFN-[gamma]/IL-4 ratio to the wild type peptide. In these
instances, a hyporesponsive T cell response to the wild-type peptide was
achieved by immunizing with an APL possessing non-conservative substitutions at
TCR contact sites, while immune deviation was accomplished using a weak-agonist
APL that retained the core determinant. Thus, certain LACK-APLs are able to
induce T cell responses with a protective phenotype in an infectious disease
such as leishmaniasis.
A biochemical and genetic study of Leishmania donovani pyruvate kinase.
Sandoval,
Will et al.
Gene, 424 (1-2), 25 (
The article relates to a biochemical and molecular biology
study of the enzyme pyruvate kinase (PYK) from the parasitic protozoa Leishmania donovani. The PYK gene was
cloned, mutagenised and over expressed and its kinetic parameters determined.
Like in other kinetoplastids, L. donovani
PYK is allosterically stimulated by the effector fructose 2,6- biphosphate and
not by fructose 1,6- biphosphate. When the putative effector binding site of L. donovani PYK was mutagenised, we
obtained two mutants with extreme kinetic behavior: Lys453Leu, which retained a
sigmoidal kinetics and was little affected by the effector; and His480Gln,
which deployed a hyperbolic kinetics that was not changed by the addition of
the effector. Molecular Dynamics (MD) studies revealed that the mutations not
only altered the effector binding site of L.
donovani PYK but also changed the folding of its domain C.
Maxicircle (mitochondrial) genome sequence
(partial) of Leishmania major: Gene
content, arrangement and composition compared with Leishmania tarentolae.
Yatawara,
Lalani et al.
Gene, 424 (1-2), 80 (
We report 8420 bp of DNA sequence data from the
maxicircle (mitochondrial) genome of Leishmania major (MHOM/SU/73/5ASKH), a
much larger portion of this genome than has been reported previously from any
Leishmania species infecting humans. This region contains 10 partial and
complete genes: 5 protein-encoding genes (COII, COIII, ND1, ND7 and Cyt b); two
ribosomal RNA subunits (12S and 9S) and three unidentified open reading frames
(MURF1, MURF4 (ATPase6) and MURF5), as in the lizard-infecting species L.
tarentolae. The genes from L. major exhibit 85-87% identity with those of L.
tarentolae at the nucleotide level and 71-94% identity at the amino acid level.
Most differences between sequences from the two species are transversions. The
gene order and arrangement within the maxicircle of L. major are similar to
those in L. tarentolae, but base composition and codon usage differ between the
species. Codons assigned for initiation for protein-coding genes available for
comparison are similar in five genes in the two species. Pre-editing was
identified in some of the protein-coding genes. Short intergenic non-coding
regions are also present in L. major as they are in L. tarentolae. Intergenic
regions between 9S rRNA and MURF5, MURF1 and ND1 genes are G+C rich and
considered to be extensive RNA editing regions. The RNA editing process is
likely to be conserved in similar pattern in L. major as in L. tarentolae.
Polyclonal hypergammaglobulinemia and high
smooth-muscle autoantibody titers with specificity against filamentous actin:
consider visceral leishmaniasis, not just autoimmune hepatitis.
Makaritsis,
Konstantinos P. et al.
International Journal of Infectious
Diseases (In Press)
Visceral leishmaniasis (VL) remains a public health problem in
most countries bordering the Mediterranean basin. Its diagnosis is challenging
and often delayed, as the main clinical picture is often indistinguishable from
that of other infectious and non-infectious diseases. Herein, we report two
unusual cases of VL that presented with several characteristics of autoimmune
hepatitis (AIH). Neither patient had a history of fever, only generalized
symptoms accompanied by polyclonal hypergammaglobulinemia, cytopenias, signs of
portal hypertension, elevated transaminases, and high titers of antinuclear and
smooth-muscle autoantibodies (SMA) with reactivity against filamentous actin
(F-actin), which has been recognized as specific to AIH. A clinical diagnosis
of AIH was considered, but a bone marrow biopsy was performed before a liver
biopsy to exclude a primary bone marrow disease. The biopsy led to the
diagnosis of VL. The diagnosis was further confirmed by IgG antibodies against
Leishmania spp. using ELISA and PCR-based assays. Treatment with amphotericin
in the first case and pentamidine in the second (because of a severe reaction
to amphotericin) was effective. From the clinical point of view, it should be
emphasized that, in cases with high titers of anti-F-actin AIH-specific SMA
accompanied by polyclonal hypergammaglobulinemia, the possibility of AIH should
be cautiously differentiated from VL; this distinction is of paramount
importance because initiation of immunosuppression for AIH treatment would be
detrimental to a patient with underlying leishmaniasis. Therefore, in such
cases and in areas where the disease is still present, it seems rational to
exclude VL before starting any immunosuppressive therapy.
Comparison of the immunomodulatory effects
of L. donovani and L. major excreted-secreted antigens,
particulate and soluble extracts and viable parasites on human dendritic cells.
Revest,
Matthieu et al.
Vaccine, 26(48),
6119 (
In an experimental model of human monocyte-derived dendritic
cells (DCs), the immunophenotype of mature DCs infected with Leishmania donovani and Leishmania major showed a weak decrease
in the cell surface expression of CD40, CD86, HLA-DR and DC-SIGN compared with
uninfected control DCs. This immunomodulatory effect was more pronounced after
stimulation with excreted-secreted antigens (ESA) of both species but absent
after stimulation with particulate and soluble extracts. Infection with viable
promastigotes, as well as stimulation with ESA from L. donovani and L. major,
decreased IL-10 and IL-12p70 secretion. To our knowledge, this is the first
direct demonstration that ESA from Leishmania promastigotes can stimulate DCs
in the same manner as viable promastigotes.
Biochemical characterization of serine
transport in Leishmania (Leishmania) amazonensis.
dos
Molecular and Biochemical Parasitology
In addition to its role as a protein component in Leishmania,
serine is also a precursor for the synthesis of both phosphatidylserine, which
is a membrane molecule involved in parasite invasion and inactivation of
macrophages, and sphingolipids, which are necessary for Leishmania to
differentiate into its infective forms. Authors have characterized serine
uptake in both promastigote and amastigote forms of Leishmania (Leishmania) amazonensis.
In promastigotes, kinetic data show a single, saturable transport system, with
a Km of 0.253 ± 0.01 mM and a maximum velocity of
0.246 ± 0.04 nmol/min per 107 cells. Serine transport increased
linearly with temperature in the range from 20 °C to 45 °C, allowing
the calculation of an activation energy of 7.09 kJ/mol. Alanine, cysteine,
glycine, threonine, valine and ethanolamine competed with the substrate at a
ten-fold excess concentration. Serine uptake was dependent on pH, with an
optimum activity at pH 7.5. The characterization of the serine transport
process in amastigotes revealed a transport system with a similar Km, energy of
activation and pH response to that found in promastigotes, suggesting that the
same transport system is active in both insect vector and mammalian host
Leishmania stages.
Leishmania manipulates sandfly feeding to
enhance its transmission.
Ready,
Paul D. et al.
Trends in Parasitology, 24(4),
151 (Apr., 2008)
Malaria parasites manipulate mosquitoes to ensure transmission
between mammalian hosts; painstaking experiments have now demonstrated that
another medically important protozoan, Leishmania, enhances its transmission
through the adaptive manipulation of one of its sandfly vectors, Lutzomyia
longipalpis. Experimental Leishmania infections specifically increased sandfly
biting persistence and feeding on multiple hosts, but only if the parasites
produced infective forms and a gel plug of filamentous proteophosphoglycan in
the anterior midgut of the sandfly. This fundamental research is relevant to
vaccine development.
Leishmania: conserved evolution - diverse
diseases.
Lynn,
Miriam A. et al.
Trends in Parasitology, 24(3), 103
(Mar., 2008)
The landmark completion of the Leishmania major genome
sequence and the recent publication of the L.
infantum and L. braziliensis
genomes revealed the surprising result that, although separated by 15-50
million years of evolution, the Leishmania genomes are highly conserved and
have less than 1% species-specific genes. Yet, these three species of
Leishmania cause distinctive and diverse diseases in humans. Here, authors
discuss these findings together with recent microarray and proteomics studies
and highlight their importance in understanding Leishmania disease phenotypes.
Polymorphisms of cpb multicopy genes in the
Leishmania (Leishmania) donovani complex.
Hide, M. et al.
Transactions of the Royal Society of
Tropical Medicine and Hygiene, 102(2), 105
(Feb., 2008)
In leishmaniasis, cysteine protease b (cpb) multicopy genes have
been extensively studied because of their implication in host-parasite
interactions. In the Leishmania donovani
complex, responsible for visceral leishmaniasis, a set of interesting
polymorphisms has been revealed, such as copy sequence or expression according
to the parasite's life stage. The single nucleotide polymorphisms observed
among these copies could be related to clinical characteristics such as
dermotropic versus viscerotropic status. cpb COOH-terminal extension (CTE) is
mainly responsible for genetic variability among the copies and appears highly
immunogenic. These results suggest that further study of the role of cpbs,
especially CTE in clinical outcome, is warranted.
Leishmania and the leishmaniases: a
parasite genetic update and advances in taxonomy, epidemiology and
pathogenicity in humans.
J.R. Baker
et al.
Advances in Parasitology, 64(1),109
(2007)
Leishmaniases remain a major public health problem today
despite the vast amount of research conducted on Leishmania pathogens. The
biological model is genetically and ecologically complex. This paper explores
the advances in Leishmania genetics and reviews population structure, taxonomy,
epidemiology and pathogenicity. Current knowledge of Leishmania genetics is
placed in the context of natural populations. Various studies have described a
clonal structure for Leishmania but recombination, pseudo-recombination and
other genetic processes have also been reported. The impact of these different
models on epidemiology and the medical aspects of leishmaniases is considered
from an evolutionary point of view. The role of these parasites in the
expression of pathogenicity in humans is also explored. It is important to
ascertain whether genetic variability of the parasites is related to the different
clinical expressions of leishmaniasis. The review aims to put current knowledge
of Leishmania and the leishmaniases in perspective and to underline priority
questions which [`]leishmaniacs' must answer in various domains: epidemiology,
population genetics, taxonomy and pathogenicity. It concludes by presenting a
number of feasible ways of responding to these questions.
The Lutzomyia
longipalpis species complex: does population sub-structure matter to
Leishmania transmission?
Maingon,
Trends in Parasitology, 24(1),12 (Jan., 2008)
Leishmania chagasi
causes visceral leishmaniasis and, to a lesser extent, atypical cutaneous
leishmaniasis in Central and
Combined diagnostic methods identify a
remarkable proportion of asymptomatic Leishmania
(Leishmania) chagasi carriers who
present modulated cytokine profiles.
de Gouvêa
Viana, Luciana et al.
Transactions of the Royal Society of
Tropical Medicine and Hygiene, 102(6), 548(June 2008)
Peripheral blood samples of 138 co-habitants from 25 families
with recently diagnosed cases of visceral leishmaniasis in the Metropolitan
Region of Belo Horizonte, Minas Gerais, Brazil, were analyzed by indirect
fluorescent antibody test (IFAT), rK39 and Leishmania
chagasi Enzyme Linked Immunosorbent Assay (ELISA), intradermal skin-test
and Polymerase Chain Reaction (PCR) over a 12-month period. The cumulative
positivity was significantly higher by PCR (29.7%) than by IFAT, rK39 ELISA, L. chagasi ELISA and intradermal
skin-test (5.1%, 6.5%, 14.5% and 2.9%, respectively). In addition, the cytokine
profile was measured in 16 of the 138 volunteers, of whom eight were
asymptomatic carriers and eight were non-infected co-habitants. The innate
immunity cells from asymptomatic carriers displayed, upon in vitro antigenic stimulation, a modulated increase in cytokine
synthesis that was distinct from that observed in non-infected volunteers. This
study suggests that the identification of a large proportion of asymptomatic
carriers is facilitated when more than one diagnostic method is applied and
that a mixed pattern of immune response is correlated with clinical status of
asymptomatic individuals. These observations suggest also that asymptomatic
infection by L. chagasi is a frequent
event and that control programs could benefit by including this indicator in
their interventions.
Genetic fingerprinting and identification
of differentially expressed genes in isolates of Leishmania donovani from Indian patients of post-kala-azar dermal
leishmaniasis.
Subba
Raju, B V et al.
Parasitology, 135(1) 23 (2008)
Post-kala-azar dermal leishmaniasis (PKDL) is an unusual
dermatosis that develops as a sequel in 5-15% of cured cases of kala-azar (KA)
after months or years of treatment in
Gene expression profiling of Leishmania (Leishmania) donovani: overcoming technical variation
and exploiting biological variation.
Decuypere,
S et al.
Parasitology, 135(2),
183 (2008)
Gene expression profiling is increasingly used in the field of
infectious diseases for characterization of host, pathogen and the nature of
their interaction. The purpose of this study was to develop a robust,
standardized method for comparative expression profiling and molecular
characterization of Leishmania donovani clinical
isolates. The limitations and possibilities associated with expression
profiling in intracellular amastigotes and promastigotes were assessed through
a series of comparative experiments in which technical and biological
parameters were scrutinized. On a technical level, our results show that it is
essential to use parasite harvesting procedures that involve minimal
disturbance of the parasite's environment in order to 'freeze' gene expression
levels instantly; this is particularly a delicate task for intracellular
amastigotes and for specific 'sensory' genes. On the biological level, authors
demonstrate that gene expression levels fluctuate during in vitro development of both intracellular amastigotes and
promastigotes. Authors chose to use expression-curves rather than single,
specific, time-point measurements to capture this biological variation.
Intracellular amastigote protocols need further refinement. A first generation
tool for high-throughput comparative molecular characterization of patients'
isolates, based on the changing expression profiles of promastigotes during in
vitro differentiation have been discussed.
Evaluation of an in vitro and in vivo model
for experimental infection with Leishmania
(Viannia) braziliensis and L. (V.) peruviana.
Gamboa, D et al.
Parasitology, 135(3), 319 (2008)
Leishmania (Viannia)
braziliensis and L. (V.) peruviana are two
parasite species characterized by a very different pathogenicity in humans
despite a high genetic similarity. Authors have previously hypothesized that L. (V.) peruviana would descend from L.
(V.) braziliensis and would have
acquired its 'peruviana' character during the southward colonization and
adaptation of the transmission cycle in the Peruvian Andes. In order to have a
first appreciation of the differences in virulence between both species, researchers
evaluated an in vitro and in vivo model for experimental
infection. A procedure was adapted to enrich culture forms in infective stages
and the purified metacyclics were used to infect macrophage cell lines and
golden hamsters. The models were tested with 2 representative strains of L. (V.) braziliensis from cutaneous and mucosal origin respectively and 2
representative strains of L. (V.) peruviana from Northern and
The health impact of polyparasitism in
humans: are we under-estimating the burden of parasitic diseases?
Pullan, R et al.
Parasitology, 135(7),
783 (2008)
Parasitic infections are widespread throughout the tropics and
sub-tropics, and infection with multiple parasite species is the norm rather
than the exception. Despite the ubiquity of polyparasitism, its public health
significance has been inadequately studied. Here available studies
investigating the nutritional and pathological consequences of multiple
infections with Plasmodium and helminth infection have been reviewed and, in
doing so, encourage a reassessment of the disease burden caused by
polyparasitism. The available evidence is conspicuously sparse but is
suggestive that multiple human parasite species may have an additive and/or
multiplicative impact on nutrition and organ pathology. Existing studies suffer
from a number of methodological limitations and adequately designed studies are
clearly necessary. Current methods of estimating the potential global morbidity
due to parasitic diseases underestimate the health impact of polyparasitism,
and possible reasons for this are presented. As international strategies to
control multiple parasite species are rolled-out, there is a number of options
to investigate the complexity of polyparasitism, and it is hoped that that the
parasitological research community will grasp the opportunity to understand
better the health of polyparasitism in humans.
A Leishmania
(L.) amazonensis ATP diphosphohydrolase
isoform and potato apyrase share epitopes: antigenicity and correlation with
disease progression.
Parasitology, 135(3), 327(2008)
A Leishmania
(Leishmania) amazonensis ATP
diphosphohydrolase isoform was partially purified from plasma membrane of
promastigotes by preparative non-denaturing polyacrylamide gel electrophoresis.
SDS-PAGE followed by Western blots developed with polyclonal anti-potato
apyrase antibodies identified diffuse bands of about 58-63 kDa, possibly
glycosylated forms of this protein. By ELISA technique, a significantly higher
total IgG antibody level against potato apyrase was found in serum from
promastigote-infected mice, as compared to the uninfected mice, confirming both
the existence of shared epitopes between the parasite and vegetable proteins,
and the parasite ATP diphosphohydrolase antigenicity. By Western blotting,
serum from amastigote-infected BALB/c mice recognizes both potato apyrase and
this antigenic ATP diphosphohydrolase isoform isolated from promastigotes,
suggesting that it is also expressed in the amastigote stage. The infection
monitored along a 90-day period in amastigote-infected mice showed reactivity
of IgG2a antibody in early steps of infection, while the disappearance of the
IgG2a response and elevation of IgG1 antibody serum levels against that shared
epitopes were associated with the progression of experimental leishmaniasis.
This is the first observation of the antigenicity of a L. (L.) amazonensis ATP
diphosphohydrolase isoform, and of the ability of cross-immunoreactivity with
potato apyrase to differentiate serologically stages of leishmaniasis in
infected mice.
Synthesis and biological evaluation of
fluorescent leishmanicidal analogues of hexadecylphosphocholine (miltefosine)
as probes of antiparasite mechanisms.
Saugar,
J.M. et al.
Journal of Medicinal Chemistry, 50(24),
5994 (
The leishmanicidal mechanism of miltefosine
(hexadecylphosphocholine, MT) is not clearly understood. Valuable insights into
its mode of action could be obtained by fluorescence techniques, given suitably
emitting analogues. In this regard, the synthesis and biological
characterization of two fully competent MT fluorescent analogues is reported
here: all-(E)-13-phenyltrideca-6,8,10,12-tetraenylphosphocholine (PTE-MT) and
all-(E)-13-phenyltrideca-8,10,12-trien-6-ynylphosphocholine (PTRI-MT). Both
compounds show large absorption coefficients and a modest, but usable,
fluorescence yield. Their activities were very similar to that of MT and were recognized
by the MT uptake system of Leishmania. Their localization in living L. donovani
promastigotes by confocal microscopy show a homogeneous intracellular
distribution of the fluorescence. The concentration of PTRI-MT within the
parasites (ca. 1.7 mM) showed a 100-fold enrichment relative to its external
concentration. These results are consistent with a multiple target
leishmanicidal mechanism for MT and validate the application of these analogues
for pharmacokinetic and diagnostic studies concerning the chemotherapy of
leishmaniasis
ANNUAL SUBSCRIPTION RATES
( For Each
Journals )
(Effective from January 2001 (Vol. 24)
Country
Subscription type Hard
Copy Hard Copy Hard Copy Hard
Copy
Category of subscriber (in Rs.) Surface
Mail (in US$) Air Mail (in US$)
A. Corporate Sector 1000 50 65 75
B. Educational & R&D 600 30 50 60
Institutions
C. Students & 400 20 40 50
Professionals
We offer a discount of 15% to those who wish to
subscribe both our journals
*
R&D TECHNOLOGY
Cutaneous leishmaniasis imported from
Delgado O
Travel Med Infect Diseases 6(6):376-9 (Nov; 2008)
Imported leishmaniasis could be defined as any case acquired
outside of a defined area in which the diagnosis of leishmaniasis is made. This
definition has been used for the diagnosis of disease in a patient who arrives
from an endemic area and displays symptoms or seeks medical attention in a
nonendemic zone. However, this phenomenon can also occur between two endemic
zones. We evaluated the epidemiologic features of imported cases of cutaneous
leishmaniasis imported from
Epidemiological aspects of cutaneous
leishmaniasis in the Iguazú falls area of
Salomón OD
et al.
Acta Trop. 2008 Oct 31.
Over the last three decades the incidence of American
cutaneous leishmaniasis (ACL) has increased sharply in
Headspace
solid phase microextraction/gas chromatography-mass spectrometry combined to
chemometric analysis for volatile organic compounds determination in canine
hair: A new tool to detect dog contamination by visceral leishmaniasis.
de Oliveira, Lidia S. et al.
Journal of Chromatography B, 875 (2), 392 (
A
new analytical methodology using HS-SPME/GC-MS was optimized in order to attain
maximum sensitivity, using multivariate strategies. The proposed method was
employed to evaluate the VOC profile exhaled from canine hair samples collected
from 8 healthy dogs and from 16 dogs infected by Leishmania infantum. 274 VOCs were detected, which could be
identified as aldehydes, ketones and hydrocarbons. After application of the
Soft Independent Modeling of Class Analogy (SIMCA) and Principal Component
Analysis (PCA) healthy and infected dogs, with similar VOCs profiles, could be
separately grouped, based on compounds such as 2-hexanone, benzaldehyde, and
2,4-nonadienal. The proposed method is non-invasive, painless, readily accepted
by dog owners and could be useful to identify several biomarkers with
applications in the diagnosis of diseases.
Leishmania (Viannia) braziliensis: New primers for
identification using polymerase chain reaction.
Marcussi, V.M. et al.
Experimental
Parasitology,
120 (4), 300 (Dec., 2008)
The
objective of this study was to develop specific primers for Leishmania (Viannia) braziliensis species identification
using PCR. The designed primers (LBF1 and LBR1) were evaluated for sensitivity
and specificity using various L. (V.)
braziliensis serodemes and various
Leishmania species and also using Trypanosoma
cruzi. A specific fragment of 536 bp was detected from 50 ng of
DNA in a crude extract derived from L. (V.)
braziliensis. The DNA fragment was
not detected when DNA from other Leishmania species or from T. cruzi was used as template in the
PCR. Furthermore, when tested with DNA from cutaneous leishmaniasis the
designed primers and reaction gave positive results. Taking into consideration
that the primers LBF1 and LBR1 could specifically identify L. (V.) braziliensis, they
could be considered for use in L.
(V.) braziliensis diagnosis and
epidemiological studies.
Canine leishmaniosis - new concepts and
insights on an expanding zoonosis: part two.
Miró,
Guadalupe et al.
Trends in Parasitology, 24(8), 371
(Aug., 2008)
Canine leishmaniasis is a widely spread zoonosis that is
potentially fatal to humans and dogs. Infection with Leishmania infantum is
considerably more prevalent than clinical disease, and infected dogs with no
signs of disease might, potentially, transmit infection. Diagnosis of
asymptomatic infection by serology is insufficient and PCR markedly increases
its sensitivity. A new therapy exclusively for canine leishmaniasis is needed
because current drugs do not reliably eliminate infection and might provoke
resistance. Protection against sand-fly bites by topical insecticides is
effective in reducing infection, and recent development of vaccines has
indicated that prevention by vaccination is feasible. Integrated prevention
with topical insecticides reducing the feeding of vectors and vaccination
blocking early infection would be the basis of successful control programme.
Leishmaniases in the mediterranean in the
era of molecular epidemiology.
Schönian,
Gabriele et al.
Trends in Parasitology, 24 (3),
135 (Mar., 2008)
Molecular tools are used increasingly for descriptive
epidemiological studies in different Mediterranean foci of visceral and
cutaneous leishmaniases. Several molecular markers with different resolution
levels have been developed to address key epidemiological questions related to
the (re-)emergence and spread of leishmaniases, as well as its risk factors:
environmental changes, immunosuppression and treatment failure. Typing and
analytical tools are improving but are not yet addressing all epidemiological
issues satisfactorily. There is an urgent need for better cooperation between
laboratory scientists and epidemiologists and for regional epidemiological
surveillance of these infectious diseases that affect all Mediterranean
countries.
Application of PCR-RFLP for the exploration
of the molecular diversity of Leishmania
infantum in
Seridi,
Nabila et al.
Transactions of the Royal Society of
Tropical Medicine and Hygiene, 102(6), 556 (June 2008)
In recent years, new methods have been developed for the
molecular typing of Leishmania that need to be extensively validated by studies
of clinical isolates in a well defined epidemiological context. The present
study is a contribution to this effort. Using PCR-RFLP of gp63 and cpb genes,
we analysed 59 isolates of L. (L.) infantum obtained from different regions
of
Dear Readers
We are happy to receive your
encouraging response to our journal. We shall, however, appreciate receiving
your critical comments and suggestions for further improvements. We are always
looking forward to your specific contributions on various aspects of the
journal. The contributions shall be duly acknowledged.
Editor
NEW LEADS
Bis-pyranobenzoquinones as a new family of
reversal agents of the multidrug resistance phenotype mediated by p-glycoprotein
in mammalian cells and the protozoan parasite leishmania.
Ravelo,
Angel G. et al.
Journal of Medicinal Chemistry 2008/10/28
Authors have synthesized a set of bis-pyranobenzoquinones
through a direct and highly efficient approach based on a double intramolecular
domino Knoevenagel hetero Diels-Alder reaction. These bis-pyranobenzoquinone
derivatives are compounds whose skeletons have similarities to those of some
anticancerous and leishmanicidal drugs. Considering that these drugs are
substrates for some members of the ATP-binding cassette (ABC) family of
proteins that confers a multidrug resistance (MDR) phenotype, we have carried
out the biological evaluation of 20 bis-pyranobenzoquinones as modulators of
the MDR phenotype in mammalian cell lines overexpressing P-glycoprotein, MRP1,
or BCRP was carried out. Moreover, we also tested some of these compounds were
tested as potential MDR modulators in a Leishmania
tropica line overexpressing a P-glycoprotein-like transporter. Compounds 9
and 10 are, in this work, the most promising reversal agents of MDR in human
cancer cell lines, while compounds 4 and 20 showed potent reversal activity of
MDR phenotype in the protozoan parasite Leishmania.
Bioinorganic chemistry of bismuth and
antimony: target sites of metallodrugs.
Ge, R. et al.
Accounts of Chemical Research, 40(4),
267 (
The biocoordination chemistry of antimony and bismuth has been
extensively investigated due to the historical use of these metals in medicine.
Structures of bismuth antiulcer agents and interactions of Bi3+ with proteins
and enzymes, such as transferrin and lactoferrin, the histidine-rich protein
Hpn, and urease, have been characterized. Sb5+ is a prodrug and is bioreduced
or activated to its active form Sb3+ intracellularly. Antimony binds to
biomolecules, such as glutathione, trypanothione, and nucleotides, and forms
binary and quartenary complexes, which may allow it to be trafficked in cells.
These studies have improved our understanding of the mechanism of action of
bismuth and antimony drugs, which in turn allows the future design of drugs
Synthesis and antiprotozoal activity of
cationic 2-phenylbenzofurans.
Bakunov,
Stanislav A et al.
Journal of Medicinal Chemistry, 51(21),
6927 (
A series of cationically substituted 2-phenylbenzofurans 1-49
have been synthesized, and their in vitro
antiprotozoal properties against Trypanosoma
brucei rhodesiense, Plasmodium
falciparum, and Leishmania donovani,
as well as cytotoxicity against mammalian cells, have been evaluated. Eight
dications exhibited antitrypanosomal activities comparable to that of
pentamidine and melarsoprol. Twenty-six compounds were more active than
pentamidine, and seven dications demonstrated increased activities against P. falciparum than artemisinin. Five
congeners were more active against L.
donovani than pentamidine. Introduction of methoxy or hydroxy groups in the
7- and/or 22-position afforded derivatives that were highly selective against
T. b. rhodesiense, P. falciparum, and
L. donovani. Fourteen
2-phenylbenzofurans displayed excellent in
vivo efficacies in the acute mouse model of trypanosomiasis, curing 3/4 or
4/4 animals at 4 × 5 mg/kg. Diamidine 1 and di(N-isopropyl)amidine 45,
administered at 4 × 1 mg/kg, exhibited potency comparable to that of
melarsoprol, providing 3/4 and 2/4 cures, respectively.
Antiprotozoal polyacetylenes from the
tanzanian medicinal plant cussonia
zimmermannii.
Senn,
Martin et al.
Journal of Natural Products, 70(10), 1565 (
From the petroleum ether extract of the root bark of Cussonia zimmermannii four
polyacetylenes, 1-4, were isolated, three of which (1-3) were active against Trypanosoma brucei rhodesiense, Trypanosoma cruzi, Plasmodium falciparum,
and Leishmania donovani.
2-(3-Aryl-3-oxopropen-1-yl)-9-tert-butyl-paullones:
A new antileishmanial chemotype.
Reichwald,
Christina et al.
Journal of Medicinal Chemistry, 51(3), 659(
A screening program directed to find new agents against Leishmania donovani, the parasite causing
visceral leishmaniasis, revealed that paullones attenuate the proliferation of
axenic amastigotes. Because these structures were not active in a test system
involving infected macrophages, a structure optimization campaign was carried
out. Concomitant introduction of an unsaturated side chain into the 2-position
and a tert-butyl substituent into the 9-position of the parent scaffold led to
compounds inhibiting also parasites dwelling in macrophages. By inclusion of
the so elaborated scaffold into a chalcone substructure, the toxicity against
uninfected host cells was significantly reduced. For the synthesis of this new
compound class, a novel modification of the Heck-type palladium-catalyzed C, C-cross
coupling strategy was used, employing a ketone Mannich base as precursor for
the alkene reactant. The so-prepared compounds exhibited improved
antileishmanial activity both on axenic amastigotes (GI50 < 1 µM) as well as
on parasites in infected macrophages.
Viridamides a and b, lipodepsipeptides with
antiprotozoal activity from the marine cyanobacterium Oscillatoria nigro-viridis.
Luke
Simmons, T et al.
Journal of Natural Products, 71(9),
1544 (
Parallel chemical and phylogenetic investigation of a marine
cyanobacterium from
Ciliatamides bioactive lipopeptides from
the deep-sea sponge Aaptos ciliate.
Nakao,
Yoichi et al.
Journal of Natural Products, 71(3), 469
(
Three lipopeptides, ciliatamides A-C (1-3), were isolated from
the deep-sea sponge Aaptos ciliata,
and their structures were elucidated on the basis of spectroscopic and chemical
methods. Ciliatamides A (1) and B (2) were found to be antileishmanial, while 2
also exhibited marginal cytotoxicity to HeLa cells.
In pursuit of natural product leads:
synthesis and biological evaluation of
2-[3-hydroxy-2-[(3-hydroxypyridine-2-carbonyl) amino] phenyl]
benzoxazole-4-carboxylic acid (a-33853) and its analogues: discovery of
n-(2-benzoxazol-2-ylphenyl) benzamides as novel antileishmanial chemotypes.
Tipparaju,
Suresh K. et al.
Journal of Medicinal Chemistry 2008/11/7
The first synthesis and biological evaluation of antibiotic 31
(A-33853) and its analogues are reported. Initial screening for inhibition of L. donovani, T. b. rhodesiense, T. cruzi,
and P. falciparum cultures followed
by determination of IC50 in L. donovani and cytotoxicity on L6 cells revealed
31 to be 3-fold more active than miltefosine, a known antileishmanial drug.
Compounds 14, 15, and 25 selectively inhibited L. donovani at nanomolar concentrations and showed much lower
cytotoxicity.
Benzoic acid derivatives from piper species
and their antiparasitic activity.
Journal of Natural Products, 71(9),
1538 (
Piper glabratum and P. acutifolium were analyzed for their
content of main secondary constituents, affording nine new benzoic acid
derivatives (1, 2, 4, 5, 7, and 10-13), in addition to four known compounds (3,
6, 8, and 9). Their structures were elucidated on the basis of spectroscopic
data. Riguera ester reactions and optical rotation measurements established the
new compounds as racemates. In the search for antiparasitic agents, the compounds
were evaluated in vitro against the
promastigote forms of Leishmania spp., Trypanosoma
cruzi, and Plasmodium falciparum.
Among the evaluated compounds, methyl
3,4-dihydroxy-5-(32-methyl-22-butenyl)benzoate (7) exhibited leishmanicidal
effect (IC50 13.8-18.5 ¼g/mL) against the three Leishmania strains used, and
methyl 3,4-dihydroxy-5-(2-hydroxy-3-methylbutenyl) benzoate (1), methyl
4-hydroxy-3-(2-hydroxy-3-methyl-3-butenyl)benzoate (3), and methyl
3,4-dihydroxy-5-(3-methyl-2-butenyl) benzoate (7) showed significant
trypanocidal activity, with IC50 values of 16.4, 15.6, and 18.5 ¼g/mL,
respectively.
pH-responsive aggregates from double
hydrophilic block copolymers carrying zwitterionic groups. Encapsulation of
antiparasitic compounds for the treatment of leishmaniasis.
Karanikolopoulos,
N. et al.
Langmuir, 23(8),
4214 (
A series of well-defined poly[(ethylene
oxide)-b-2-(dimethylamino)ethyl methacrylate] (PEO-b-PDMAEMA) diblock
copolymers were synthesized by atom transfer radical polymerization (ATRP)
techniques. Post-polymerization reactions were performed to transform a portion
of the tertiary amine groups of the PDMAMA into phosphoro-zwitterions.
Antiparasitic drugs used for the treatment of Leishmania were incorporated into
the copolymer aggregates. The effect of the solution pH, the zwitterion
content, temperature, and the quantity of the incorporated drug on the
aggregation behavior of the copolymers was tested.
Hydrolytic reactivity trends among
potential prodrugs of the O2-glycosylated diazeniumdiolate family.
Targeting nitric oxide to macrophages for antileishmanial activity.
Valdez,
Carlos A. et al.
Journal of Medicinal Chemistry, 51(13),
3961 (
Glycosylated diazeniumdiolates of structure R2NN(O)NO-R2
(R2 = a saccharide residue) are potential prodrugs of the nitric
oxide (NO)-releasing but acid-sensitive R2NN(O)NO-ion.
Moreover, cleaving the acid-stable glycosides under alkaline conditions
provides a convenient protecting group strategy for diazeniumdiolate ions. In
the study authors report comparative
hydrolysis rate data for five representative glycosylated diazenium-diolates at
pH 14, 7.4, and 3.8-4.6 as background for further developing both the
protecting group application and the ability to target NO pharmacologically to
macrophages harboring intracellular pathogens. Confirming the potential in the
latter application, adding R2NN(O)NO-GlcNAc (where R2N =
diethylamino or pyrrolidin-l-yl and GlcNAc = N-acetylglucosamin-l-yl) to
cultures of infected mouse macrophages that were deficient in inducible NO
synthase caused rapid death of the intracellular protozoan parasite Leishmania major with no host cell
toxicity.
Synthesis and in vitro antiprotozoal activities of water-soluble, inexpensive 3, 7-bis
(dialkylamino) phenoxazin-5-ium derivatives.
Ge,
Jian-Feng et al.
Journal of Medicinal Chemistry, 51(12),
3654 (
3,7-Bis(dialkylamino)phenoxazinium salts were synthesized and
evaluated for in vitro activities
against Plasmodium falciparum, Trypanosoma cruzi, T. brucei rhodesiense, and Leishmania
donovani. Notably, the compounds showed potent antiprotozoal activities,
especially against P. falciparum and T. cruzi. The compounds with alkyl side
chains less than three carbons in length possessed good activities with high
selective indices.
Selection of the most promising
2-substituted quinoline as antileishmanial candidate for clinical trials.
Vieira,
Nashira Campos et al.
Biomedicine & Pharmacotherapy, 62(10),
684 (Dec., 2008)
The antileishmanial evaluation of more than one hundred
2-substituted quinolines led us to identify three compounds for further
studies: compound 1 (2-N-propylquinoline), compound 2 (2-(2-methoxyethenyl)quinoline)
and compound 3 (2-(2-hydroxyprop-2-enyl)quinoline). The final selection of a
potential drug candidate was mainly based on chemical stability and acute oral
toxicity as discriminating criteria. The most stable compound in various
conditions was 2-N-propylquinoline (compound 1). Only reversible toxicity signs
were observed for compound 1 at 1000 mg/kg after a treatment by oral route
at a single dose and no sign was detected at 100 mg/kg. Interestingly,
2-substituted quinolines were active on a Leishmania
donovani line, resistant to sitamaquine, a 8-aminoquinoline, suggesting
that 2-substituted quinolines and 8-aminoquinoline probably affect a different
target in L. donovani.
Synthesis and evaluation of antiparasitic
activities of new 4-[5-(4-phenoxyphenyl)-2h-pyrazol-3-yl] morpho-line
derivatives.
Kuettel,
S. et al.
Journal of Medicinal Chemistry, 50(23),
5833 (
A series of new
4-[5-(4-phenoxyphenyl)-2H-pyrazol-3-yl]morpholine derivatives, prepared by two
synthetic routes, were in vitro
assayed against three Trypanosoma strains,
Leishmania donovani, and Plasmodium falciparum K1. Seven out of
17 compounds showed moderate to very good activity against blood stage T. b. rhodesiense, with 10 and 17
exhibiting highest potency (IC50 of 1.0 and 1.1 M, respectively).
Interestingly, the -diketone precursors 1-3 had good antitrypanosomal activity
toward the insect stage, with IC50 values of 1.0-3.4 M. Among different
compounds with moderate activity against T.
cruzi, compound 17 showed the lowest IC50 value of 9.5 M; thus, the series
seemed to act selectively toward the different Trypanosoma parasites. Eight
compounds were moderately active against L.
donovani, with 2, 3, and 12 being the most promising ones (IC50 values of
2.3-5.2 M), whereas compound 14 was the only derivative with good activity
against P. falciparum (IC50 of 3.7
M).
Synthesis, DNA binding, and leishmania
topoisomerase inhibition activities of a novel series of
anthra[1,2-d]imidazole-6,11-dione derivatives.
Chaudhuri,
P. et al.
Journal of Medicinal Chemistry, 50(10),
2536 (
Nine novel anthrax [1,2-d]imidazole-6,11-diones, differing in
their side chain, were synthesized. UV-vis spectroscopy and viscometric
titrations of these molecules with duplex DNA were used to assess their binding
with DNA. Five of the nine compounds showed high inhibition activity against
topoisomerase I of Leishmania donovani,
with the one bearing the tetrazole side chain exhibiting an IC50 ~ 1 M. The
inhibition activities were not related with their DNA binding affinity and
depended on the nature of the side chain.
Gift Subscription
Contributions are
invited for our Journals:( I ). Current R&D Highlights and
( ii.) Industry
Highlights on any of the following aspects of
Drugs &
Pharmaceuticals
Industry and
Policy
R&D and
Technology
Biotechnology
Health Care
Natural Products
Traditional
Medicines
Information
Technology
Intellectual
Property Rights
Diagnostics &
Prophylactics
You may
contribute detailed Feature articles (upto 2000 words) or your views (upto 1000 words) on any of the above or
related topics concerning Drugs and Pharmaceuticals. All those, whose articles
or views are accepted for publication, will be offered one year Gift
Subscription for one of the above journals.
Kindly send your
article(s) entered in a floppy in MS-Word, Wordstar, Word Perfect, Pagemaker,
or in non document mode alongwith a print out as per the Instructions to
Authors given on the Back-inside cover.
For Further
details, please contact:
The Scientist
–Charge
Documentation
& Library Services
Central Drug
Research Institute
Post Box No.173,
Email SIRNET: root%[email protected]
INTERNET:
[email protected]
Tel: 2613812:
2612411-18 (Extn. 4276)
Fax: 0522-2623405
& 2623938
Web site:
www.cdriindia.org
NATURAL PRODUCTS
Antiparasitic, antineuroinflammatory, and
cytotoxic polyketides from the marine sponge Plakortis angulospiculatus collected in
Kossuga,
Miriam H. et al.
Journal of Natural Products, 71(3),
334 (
Investigation of the bioactive crude extract from the sponge Plakortis angulospiculatus from
Trypanocidal and antileishmanial
dihydrochelerythrine derivatives from Garcinia
lucida.
Fotie,
Jean et al.
Journal of Natural Products, 70(10),
1650 (
Three benzo[c] phenanthridine alkaloids have been isolated
from the stem bark of Garcinia lucida:
dihydrochelerythrine (1), 6-acetonyldihydrochelerythrine (2), and its new
derivative, (S)13-(9,10-dihydro-22,32-di-hydroxy-7,8-dimethoxy-10-methyl-1,2-benzo-phenanthridin-9-yl)
propan-23-one (luci-damine A) (3). The new diisoprenylated derivative of
lucidamine B (4) was obtained by semisynthesis. These dihydrochelerythrine
derivatives as well as the crude extract displayed attractive antiprotozoal
activity against Trypanosoma brucei
and Leishmania donovani, with little
toxicity to Vero cells and the host cells. This is the first trypanocidal and
antileishmanial bioguided study of G.
lucida, and the activity of the crude extract as well as of the
dihydrochelerythrine derivatives are reported for the first time.
Hydroxyalkenylresorcinols from Stylogyne turbacensis.
Jimenez-Romero,
C. et al.
Journal of Natural Products, 70(8),
1249 (
Two new compounds,
5-(11'(S)-hydroxy-8'-heptadecenyl)resorcinol (3) and
5-(12'(S)-hydroxy-8',14'-heptadecadienyl)resorcinol (4), were isolated from the
leaves of Stylogyne turbacensis
together with the known analogue metabolites 1 and 2. Compounds 3 and 4 showed
the strongest activity in the leishmania assay, 7 and 3 M, respectively, while
compounds 1, 2, and 4 showed moderate activity against a drug-resistant strain
of Trypanosoma cruzi with IC50 values
of 30, 25, and 22 M, respectively. Additional testing in MCF-7 and NCI-H460 was
performed for compounds 3 and 4. The structures of compounds 1-4 were
elucidated using NMR, MS, and other spectroscopic data. The absolute
stereochemistry of compounds 3 and 4 was also investigated using the Mosher ester
approach. Peracetylated derivatives of these four metabolites were produced and
their activities determined in the Trypanosoma
cruzi assay.
Bioactive-pregnane-type steroidal alkaloids
from Sarcococca hookeriana.
Devkota,
Journal of Natural Products, 71(8), 1481 (
The bioassay-guided phytochemical investigation of Sarcococca hookeriana with respect to
cholinesterase inhibitory properties has yielded two new 5±-pregnane-type
steroidal alkaloids, hookerianamides J (1) and K (2), along with eight known
compounds (3-10). The structures of 1 and 2 were elucidated by spectroscopic
methods. These compounds displayed good to moderate activities in vitro against the enzymes
acetylcholinesterase (IC50 8.1-48.5 ¼M) and butyrylcholinesterase (IC50 0.4-4.0
¼M). Compounds 1-10 were also tested in
vitro for their leishmanicidal activity against Leishmania major and for their antibacterial activities against
Bacillus subtilis, Micrococcus luteus, Streptococcus faecalis, and Pseudomonas pallida.
Limonoid orthoacetates
and antiprotozoal compounds from the roots of Pseudocedrela kotschyi.
Hay, A.-E. et al.
Journal of
Natural Products, 70(1), 9 (
The dicholoromethane extract of Pseudocedrela kotschyi root demonstrated marked antileishmanial
properties in preliminary screening of extracts from 21 species commonly used
in Malian traditional medicine. Phytochemical investigation of the active
extract yielded three novel phragmalin-type limonoid orthoacetates (1-3), named
kotschyins A-C, and the known compounds 7-deacetylgedunin (4) and
7-deacetyl-7-oxogedunin (5). The structures of 1-3 were elucidated by
analytical methods including 1D- and 2D-NMR spectroscopy together with MS
spectroscopy. The relative configurations of 1-3 were assigned on the basis of
NOE correlations. The extract and the isolated compounds were tested for their
antiprotozoal activities against Leishmania
donovani, Trypanosoma brucei
rhodesiense, Trypano-soma cruzi, and Plasmodium falciparum as well as for cytotoxicity
toward the L-6 cell line. The crude extract and the two gedunin derivatives
exhibited good in vitro activity
against all of these parasites.
Cycloartane Triterpenoids from Astragalus bicuspis.
Choudhary,
M. Iqbal et al.
Journal of Natural Products, 71(9),
1557 (
Three new cycloartane glycosides have been isolated from Astragalus bicuspis. They were
identified as
6±-hydroxy-3-O-²-xylopyranosyloxy-24,25,26,27-tetranor-9,19-cyclolanosta-16,23-lactone
(1), 6± -hydroxy-23-methoxy-16²,23(R)-epoxy-24,25,26,27-tetranor-9,19-cyclolanosta-3-O-²-xyloside
(2), and 23(R), 24(S),25(R), 26(S)-16²/23, 23/26, 24/25-triepoxy-6±, 26-dihydroxy-9,
19-cyclo-lanosta-3-O-²-xyloside (3), on the basis of their spectroscopic data.
Two known cycloartane derivatives, 4 and 5, were also obtained from this plant.
Compounds 2-5 were tested for leishmanicidal activity against Leishmania major promastigotes and for
cytotoxicity against 3T3 cancer cells.
Antiprotozoal activities of
heterocyclic-substituted xanthones from the marine-derived fungus chaetomium
sp.
Pontius,
Alexander et al.
Journal of Natural Products, 71(9),
1579 (
Investigations of the marine-derived fungus Chaetomium sp. led to the isolation of
the new natural products chaetoxanthones A, B, and C (1-3). Compounds 1 and 2
are substituted with a dioxane/tetrahydropyran moiety rarely found in natural
products. Compound 3 was identified as a chlorinated xanthone substituted with
a tetrahydropyran ring. The configurational analysis of these compounds
employed CD spectroscopy, modified Mosher’s method, and selective NOE gradient
measurements. Compound 2 showed selective activity against Plasmodium falciparum with an IC50 value of 0.5 ¼g/mL without being
cytotoxic toward cultured eukaryotic cells. Compound 3 displayed a moderate
activity against Trypanosoma cruzi
with an IC50 value of 1.5 ¼g/mL.
Concise synthesis of a pentasaccharide
related to the anti-leishmanial triterpenoid saponin isolated from Maesa balansae.
Rajput,
Vishal Kumar et al.
The Journal of Organic Chemistry, 73(17),
6924 (
Concise synthesis of the glycone part (a pentasaccharide) of
the anti-leishmanial triterpenoid saponin isolated from Maesa balansae is
reported. A late-stage TEMPO-mediated oxidation of a primary hydroxyl group to
carboxylic acid has been achieved under phase-transfer conditions. Glyco-sylations
were performed either by thioglycoside or glycosyl trichloroacetimidate
activation using sulfuric acid immobilized on silica (H2SO4-silica)
in conjunction with N-iodosuccinimide and alone, respectively. H2SO4-silica
was proved to be a better choice as promoter than conventional Lewis acid
promoters such as TfOH or TMSOTf.
Antileishmanial compounds from Cordia fragrantissima collected in
Mori,
Kanami et al.
Journal of Natural Products, 71(1), 18
(
A methanol extract of the wood of Cordia fragrantissima, collected in
Content, composition, and bioactivity of
the essential oils of three basil genotypes as a function of harvesting.
Zheljazkov,
Valtcho D. et al.
Journal of Agricultural and Food
Chemistry, 56(2), 380 (
A study was conducted to evaluate the effect of cut on biomass
productivity, oil content, composition, and bioactivity of Ocimum basilicum L. (cvs. German and Mesten) and Ocimum sanctum L. (syn. O. tenuiflorum
L.) (cv. Local) in
Mitracarpus
frigidus aerial parts exhibited potent antimicrobial, antileishmanial, and
antioxidant effects
Fabri,
R.L. et al.
Bioresource Technology, 100(1), 428
(Jan., 2009)
The crude extract and the hexane, CH2Cl2,
EtOAc, n-BuOH, and hydromethanolic fractions of the aerial parts of Mitracarpus frigidus were evaluated
against promastigote forms of two species of Leishmania (L. chagasi and L.
amazonensis), 11 strains of bacteria (Staphylococcus aureus, Pseudomonas
aeruginosa, Salmonella enterica sorovar Tythimurium, Shigella sonnei,
Klebsiella pneumoniae, Escherichia coli, Micrococcus luteus, Enterococcus
faecalis, Enterobacter cloacae, Streptococcus pyogenes and Bacillus cereus) and
two yeasts (Candida albicans and Cryptococcus neoformans). The antioxidant
activity (DPPH radical scavenging activity and reducing power), cytotoxicity
against mammalian cells, and the contents of phenolics and flavonoids were determined.
Phytochemical analysis of the major groups of phytoconstituents is also
reported. All samples showed antioxidant activity which was positively
correlated to the content of phenolic compounds. S. sonnei, B. cereus and C.
neoformans were susceptible to all extracts tested, except for the n-BuOH and
hydromethanolic fractions, which demonstrated no antimicrobial activity. The
lowest MIC was recorded for the CH2Cl2 fraction against
C. neoformans (MIC of 10 [mu]g/ml), followed by B. cereus, S. sonnei, and E.
cloacae (MIC of 20, 39 and 39 [mu]g/ml, respectively). The CH2Cl2
fraction was the most effective against L. chagasi (IC50 of 6.7 [mu]g/ml),
and the hydromethanolic fraction exhibited the best activity against L.
amazonensis (IC50 of 9 [mu]g/ml). A cytotoxic effect on mammalian cells
was observed only for the crude extract and CH2Cl2
fraction at the concentrations of 130 and 31 [mu]g/ml, respectively. These
results suggest that M. frigidus has
interesting antimicrobial, antileishmanial and antioxidant activities.
Antiplasmodial and leishmanicidal activity
of biflavonoids from Indian Selaginella
bryopteris.
Kunert,
Olaf et al.
Phytochemistry Letters, 1(4), 171
(
A series of eleven biflavonoids containing amentoflavone and
hinokiflavone derivatives from the Indian medicinal herb Selaginella bryopteris has been investigated for their
antiprotozoal activity using in vitro
assays against the K1 strain of Plasmodium
falciparum, Leishmania donovani, Trypanosoma
brucei rhodesiense and Trypanosoma
cruzi. The highest antiprotozoal activity was displayed by
7,4',7''-tri-O-methylamentoflavone which exhibited an IC50 of 0.26 [mu]M.
This compound showed no significant cytotoxicity
(IC50 > 150 [mu]M) evaluated using L-6 cells. The strongest
activity against Leishmania was detected for 2,3-dihydrohinokiflavone
(IC50 = 1.6 [mu]M), whereas for Trypanosoma no significant
activity was observed (IC50 > 12.5 [mu]g/mL for the extract).
To evaluate the in vivo activity against Plasmodium of the most active compound,
trimethylated amentoflavones were obtained by partial synthesis starting from
amentoflavone. The synthesized mixture of trimethylated amentoflavones did not
show activity in the Plasmodium berghei
mouse model against female NMRI mice at 50 mg/kg.
Pumilanol, an antiprotozoal isoflavanol
from Tephrosia pumila.
Ganapaty,
Seru et al.
Phytochemistry Letters, 1(4), 175
(
A novel isoflavan-4-ol (pumilanol),
(rel)-3[beta]-(2'-hydroxy-4',5'-methylenedioxyphenyl)-6-methoxy-4[alpha],7-dihydroxybenzo-3,4-dihydropyran
(1) and two known flavonoids, tephrinone (2) and rotenone (3) together with
lupeol and stigmasterol were isolated from the roots of Tephrosia pumila (Fabaceae) from a collection made in Andhra
Pradesh, India. The structures of the compounds were determined by MS, 1D and
2D-NMR spectral analysis. Pumilanol (1) exhibited significant antiprotozoal
activity against T. b. rhodensiense, T. cruzi and L. donovani with IC50 of 3.7, 3.35 and 17.2 [mu]g/mL,
respectively, but displayed high toxicity towards L-6 (IC50 of
17.12 [mu]g/mL) rat skeletal myoblasts.
Immunomodulatory pretreatment with Kalanchoe pinnata extract and its
quercitrin flavonoid effectively protects mice against fatal anaphylactic shock
Cruz, E.A et al.
International Immunopharmacology, 8(12),. 1616 (
The immunosuppressive action of the aqueous extract of Kalanchoe pinnata (Kp) in mice is
reported in literature, the present study, reports on the protective effect of
Kp in fatal anaphylactic shock, likewise a Th2-driven immunopathology, and the
identification of its active component. Mice daily treated with oral Kp during
hypersensitization with ovalbumin were all protected against death when
challenged with the allergen, as compared with the 100% mortality in the
untreated group. A single intraperitoneal dose 3 h prior to challenge was
partially effective. Oral protection was accompanied by a reduced production of
OVA-specific IgE antibodies, reduced eosinophilia, and impaired production of
the IL-5, IL-10 and TNF-[alpha] cytokines. In vitro, Kp prevented
antigen-induced mast cell degranulation and histamine release. Oral treatment
with the quercitrin flavonoid isolated from Kp prevented fatal anaphylaxis in
75% of the animals. These findings indicate that oral treatment with Kp
effectively downmodulates pro-anaphylactic inducing immune responses.
Protection achieved with quercitrin, although not maximal, suggests that this
flavonoid is a critical component of Kp extract against this extreme allergic
reaction.
Effect of Brazilian copaiba oils on Leishmania amazonensis.
Santos,
Adriana O. et al.
Journal of Ethnopharmacology, 120 (2),
204(
Copaiba oil has been used in folk medicine since the 19th
century. The use of copaiba oils to treat leishmaniasis is cited in several
ethnopharmacological studies. Nevertheless, the potential antileishmania of
copaiba oils had not been studied. Eight different kinds of Brazilian copaiba
oils were screened for antileishmanial activity. The antiproliferative effect of
copaiba oil on promastigote and amastigote axenic were determined. To determine
the survival index peritoneal macrophage were infected with promastigotes of Leishmania amazonensis and treated with
copaiba oil. The cytotoxic effect of copaiba oil was assessed on macrophage
strain J774G8 by assay of sulforhodamine B.Copaiba oils showed variable levels
of activity against promastigote forms with IC50 values in the range between 5
and 22 [mu]g/mL. The most active oil was that from Copaifera reticulata with IC50 values of 5, 15, and
20 [mu]g/mL for promastigote, axenic amastigote and intracellular
amastigote forms, respectively. Amphotericin B showed IC50 of 0.058 and
0.231 [mu]g/mL against promastigote and amastigote forms, respectively.
Cytotoxicity assay showed that this copaiba oil obtained from Copaifera reticulata showed low
cytotoxicity against J774G8 macrophages.
Alkaloids: Future prospective to combat
leishmaniasis.
Mishra BB et al.
Fitoterpia, 2008 Oct 31
Leishmaniasis, a vector-borne parasitic disease resulting from
infection of macrophages by obligate intracellular parasites of genus
Leishmania, has been considered a major tropical disease by the World Health
Organization. Generic pentavalent antimonials have been the mainstay for
therapy in the endemic regions because of its efficacy and cost effectiveness.
However, the growing incidence of resistance for the pentavalent antimony
complex in endemic and non-endemic regions has seriously hampered their use in
these regions. The second line drugs such as amphotericin B, paromomycin and
miltefosine are the other alternatives, but they merely fulfill the desired
requirements of a safe drug. The recent researches focused on plants have shown
a wise way to get a true and potentially rich source of drug candidates against
leishmaniasis, where alkaloids have been found more effective. The present
review initially highlights the current status of leishmaniasis, synergy of the
disease with HIV, therapeutic options available and in later sections
summarizes all alkaloids, which have shown significant antileishmanial
activities
Leishmanicidal activity of Yucatecan
medicinal plants on Leishmania species responsible for cutaneous leishmaniasis.
Getti
G et al.
J. Parasitology, 2008 Sep 4:1.
The leishmanicidal activity of 15 extracts and 4 pure
metabolites obtained from Urechites
andrieuxii, Colubrina greggii, Dorstenia contrajerva, and Tridax procumbens was evaluated using
the newly developed MTS
({3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl}-2H-tetrazolium,
inner salt) assay, optimized for promastigotes of Leishmania major, L. tropica, and L. aethiopica, as well as for L.
aethiopica axenic amastigotes. The assay was then used for calculating the
percentage of viable stationary phase parasites after a 24 hr treatment with
each plant extract or pure metabolite. The 3 most active samples, 2 from C. greggii (NCG-5C and DCG-3A) and 1
from T. procumbens (TPZ-2A) showed
LD50 values of 18.5, 7.2, and 62.4 µg/ml, respectively, on stationary promastigotes,
and of 95.2, 27.1, and 94.2 microg/ml, on amastigotes of L. aethiopica. Moreover, TPZ-2A and DCG-3A also significantly
reduced the percentage of infected monocyte-derived macrophages (THP-1). The
percentage of infected cells went from 69.9 % (+/-2.5), to 20.8 % (+/-2) when
treated with the DCG-3A fraction and to 14.9 % (+/-0.5) when treated with
TPZ-2A, without significantly decreasing the number of human cells. These
findings indicate the presence of potentially bioactive metabolites in the roots
of C. greggii and in T. procumbens and reflect the importance
of pursuing the bioassay-guided purification of these metabolites.
Altenusin,
a biphenyl isolated from the endophytic fungus Alternaria sp., inhibits trypanothione reductase from Trypanosoma cruzi
Betania
Barros Cota et al.
FEMS Microbiol Lett. 2008,
Parasitic protozoan species belonging to the genera Trypanosoma and Leishmania
are the etiological agents of several diseases in tropical areas of the world,
for which there is an urgent need for effective and affordable treatment. In
this regard, we are screening the Brazilian biodiversity, especially its flora
and mycota, for natural products that could serve as leads for drug development
against these diseases. Trypanothione reductase (TR) is an enzyme involved in
the protection of Trypanosoma
and Leishmania species against
oxidative stress, and is considered to be a validated drug target. The
endophytic fungus Alternaria
sp. (UFMGCB55) was isolated from the plant Trixis
vauthieri DC (Asteraceae),
known to contain trypanocidal compounds. The organic extract of the culture of Alternaria sp. was able to inhibit
TR by 99%, when tested at 20 μg mL−1.
Fractionation of the extract identified altenusin, a biphenyl derivative with
an IC50 value of 4.3±0.3 μM in the TR assay. This compound
is the first in its class to have shown TR inhibitory activity, opening new
perspectives for the design of more effective derivatives that could serve as
drug leads for new chemotherapeutic agents to treat trypanosomiasis and
leishmaniasis.
Brazilian
flora extracts as source of novel antileishmanial and antifungal compounds.
Tempone AG et
al.
Mem inst. Oswaldo Cruz, 2008 Aug;103(5):443-9.
Natural products have long been providing important drug leads
for infectious diseases. Leishmaniasis is a protozoan parasitic disease found
mainly in developing countries, and it has toxic therapies with few
alternatives. Fungal infections have been the main cause of death in
immunocompromised patients and new drugs are urgently needed. In this work, a
total of 16 plant species belonging to 11 families, selected on an ethnopharmacological
basis, were analyzed in vitro against Leishmania
(L.) chagasi, Leishmania (L.) amazonensis,
Candida krusei, and C. parapsilosis. Of these plant species,
seven showed antifungal activity against C.
krusei, five showed antileishmanial activity against L. chagasi and four against L.
amazonensis, among them species of genus Plectranthus. Our findings confirm
the traditional therapeutic use of these plants in the treatment of infectious
and inflammatory disorders and also offer insights into the isolation of active
and novel drug prototypes, especially those used against neglected diseases as
Leishmaniasis.
Evaluation of the influence of tissue
parasite density on hematological and phenotypic cellular parameters of
circulating leukocytes and splenocytes during ongoing canine visceral leish-maniasis.
Guerra LL et al.
Parasitol Res. 2008 Nov 5.
During Leishmania infection, tissue parasitism at different
sites may differ and imply distinct immunopathological patterns during canine
visceral leishmaniasis (CVL). For this reason, authors have assessed by flow
cytometry the impact of spleen and skin parasite density on the phenotypic
profile of splenocytes and circulating leukocytes of 40 Brazilian dogs
naturally infected by Leishmania chagasi
categorized according to splenic and cutaneous parasite load. Our major
statistically significant findings demonstrated that dogs with splenic high
parasitism presented a significant decrease in absolute counts of CD5(+) T
lymphocytes in comparison with dogs presenting splenic medium
parasitism.Moreover, a decrease in the absolute number of circulating monocytes
was observed as a hallmark of high parasitism. The increased frequency of
CD8(+) T cells is associated with low splenic parasitism during CVL. Although
we did not found any significant differences between the immunophenotypic
analysis performed in circulating lymphocytes according to cutaneous parasite
load, there were negative correlations between CD5(+) and CD8(+) T cells and
cutaneous parasite density reemphasizes the role of T cell-mediated immune
response in resistance mechanisms during ongoing CVL. These results add new
insights about the pathogenesis of CVL and may help in the establishment of
additional tools for future studies on drugs and vaccine approaches.
Seroprevalence of visceral leishmaniasis in
stray dogs in kocaeli.
Tamer GS et al.
Turkiye Parazitol Derg. 2008;32(3):183-6.
Dogs are the most important reservoir of visceral
leishmaniasis (VL). A male child who lives in Köseköy in Kocaeli was diagnosed
with VL. Since this child had never been outside Kocaeli, serum samples of 65
stray dogs were analyzed for canine VL using the indirect fluorescence antibody
test (IFAT) and ELISA. Two dogs (3.07%) tested positive with both ELISA and
IFAT. Leishmania amastigotes were observed in the lymph aspiration material
from one of them. Growth was observed in NNN medium inoculated with lymph
aspiration material from the other dog. This was the first study investigating
canine VL prevalence in our city and plans were made for control of the
disease.
Identification of highly specific and
cross-reactive antigens of leishmania species by antibodies from Leishmania (leishmania) chagasi naturally infected dogs.
Vale AM et al.
Zoonoses Public Health. 2008 Nov 1.
The Leishmania species present a genetic homology that ranges
from 69 to 90%.Because of this homology, heterologous antigens have been used
in the immunodiagnosis and vaccine development against Leishmania infections.
In the current work, we describe the identification of species-specific and
cross-reactive antigens among several New World Leishmania species, using
symptomatic and asymptomatic naturally Leishmania
chagasi-infected dog sera. Soluble antigens from five strains of New World
Leishmania were separated by electrophoresis in SDS-PAGE and immunoblotted.
Different proteins were uniquely recognized in the L. chagasi panel by either symptomatic or asymptomatic dog sera
suggesting their use as markers for the progression of disease and diagnosis of
the initial (sub-clinical) phase of the infection. Cross-reactive antigens were
identified using heterologous antigenic panels (L. amazonensis strains PH8 and BH6, L. guyanensis and L.
braziliensis). L. guyanensis
panel showed the highest cross-reactivity against L. chagasi specific antibodies, suggesting that proteins from this
extract might be suitable for the diagnosis of visceral canine leishmaniasis.
Interestingly, the 51 and 97 kDa proteins of Leishmania were widely recognized
(77.8% to 100%) among all antigenic panels tested, supporting their potential
use for immunodiagnosis. Finally, authors identified several leishmanial
antigens that might be useful for routine diagnosis and seroepidemiological
studies of the visceral canine leishmaniasis.
Transmission potential, skin inflammatory
response, and parasitism of symptomatic and asymptomatic dogs with visceral
leishmaniasis.
Vercosa BB
et al.
BMC Vet Res. 2008 Nov 6;4(1):45. Visceral leishmaniasis in
Discovery of novel vaccine candidates and
drug targets against visceral leishmaniasis using proteomics and
transcriptomics.
Kumari S et al.
Curr Drug Targets. 2008 Nov;9(11):938-47.
Among the three clinical forms (cutaneous, mucosal and
visceral) of leishmaniasis visceral (VL) one is the most devastating type
caused by the invasion of the reticuloendothelial system of human by Leishmania donovani, L. infantum and L.chagasi.
A glutathione-specific aldose reductase of Leishmania donovani and its potential
implications for methylglyoxal detoxification pathway.
Rath J et al.
Gene. 2008 Oct 15.
Methylglyoxal is mainly
catabolized by two major enzymatic pathways. The first is the ubiquitous
detoxification pathway, and the second is the glyoxalase pathway. In addition
to the glyoxalase enzymes, aldose reductases also play a crucial role in
lowering the levels of methylglyoxal. The gene encoding aldose reductase (ALR)
has been cloned from Leishmania donovani, a protozoan parasite causing visceral
leishmaniasis. DNA sequence analysis revealed an open reading frame (ORF) of
~855 bp encoding a putative protein of 284 amino acids with a calculated
molecular mass of 31.7 kDa and a predicted isoelectric point of 5.85. The
sequence identity between L. donovani ALR (LdALR) and mammals and plants is
only 36-44%. The ORF is a single copy gene. A protein with a molecular mass
that matched the estimated ~74 kDa according to the amino acid composition of
LdALR with a maltose binding tag present at its N-terminal end was induced by
heterologous expression of LdALR in Escherichia
coli. In the presence of glutathione, recombinant LdALR reduced
methylglyoxal with a K(m) of approximately 112 muM. Comparative structural
analysis of the human ALR structure with LdALR model suggests that the active
site anchoring the N-terminal end of the glutathione is highly conserved.
However, the C-terminal end of the glutathione backbone is expected to be
exposed in LdALR, as the residues anchoring the C-terminal end of the
glutathione backbone come from the three loop regions in human, which are
apparently shortened in the LdALR structure. Thus, the computational analysis
provides clues about the expected mode of glutathione binding and its interactions
with the protein. This is the first report of the role of an ALR in the
metabolic disposal of methylglyoxal in L.
donovani and of thiol binding to a kinetoplastid aldose reductase.
Vaccination with a novel recombinant
Leishmania antigen plus MPL provides partial protection against L. donovani
challenge in experimental model of visceral leishmaniasis.
Bhardwaj S
et al.
Exp Parasitol. 2008 Oct 17.
The acquisition of immunity following subclinical or resolved
infection with the intracellular parasite Leishmania donovani suggests that
vaccination could prevent visceral leishmaniasis. The characteristics and in vitro stimulating capability of the
recombinant proteins expressed by previously identified clones on the basis of
their capacity to stimulate an indigenously established Leishmania-specific
cell line leading to high level of IFN-gamma suggested these to be potential
candidates for immunoprophylaxis against leishmaniasis. In this study, we
investigated the protective efficacy of purified recombinant proteins from two
of the identified cDNA clones along with the adjuvant MPL, in a hamster model
of experimental leishmaniasis. Authors demonstrate here that the immunization
of animals with one of the recombinant proteins (rF14) having 97% similarity to
C1 clone of L. chagasi ribosomal
protein gene P0 (rLiP0) along with MPL provided partial protection against the
virulent challenge of L. donovani. The
absence of antigen-specific lymphoproliferative responses in these immunized
animals may be responsible for the lack of complete and long-lasting
protection.
Prevalence of antibodies and cell mediated
immune response against Leishmania major
in feral nonhuman primates from
Gicheru MM
et al.
Acta Trop. 2008 Oct 15.
In
DNA based vaccination with a cocktail of
plasmids encoding immunodominant Leishmania
(Leishmania) major antigens
confers full protection in BALB/c mice.
Ahmed SB et al.
Vaccine. 2008 Oct 22.
Despite the lack of effective vaccines against parasitic
diseases, the prospects of developing a vaccine against leishmaniasis are still
high. With this objective, authors have tested four DNA based candidate
vaccines encoding to immunodominant leishmania antigens (LACKp24, TSA, LmSTI1
and CPa). These candidates have been previously reported as capable of
eliciting at least partial protections in the BALB/c mice model of experimental
cutaneous leishmaniasis. When tested under similar experimental conditions, all
of them were able to induce similar partial protective effects, but none could
induce a full protection. In order to improve the level of protection we have
explored the approach of DNA based vaccination with different cocktails of
plasmids encoding to the different immunodominant Leishmania antigens. A
substantial increase of protection was achieved when the cocktail is composed
of all of the four antigens; however, no full protection was achieved when mice
were challenged with a high dose of parasite in their hind footpad. The full
protection was only achieved after a challenge with a low parasitic dose in the
dermis of the ear. It was difficult to determine clear protection correlates,
other than the mixture of immunogens induced specific Th1 immune responses
against each component. Therefore, such an association of antigens increased
the number of targeted epitopes by the immune system with the prospects that
the responses are at least additive if not synergistic. Even though, any
extrapolation of this approach when applied to other animal or human models is
rather hazardous, it undoubtedly increases the hopes of developing an effective
leishmania vaccine.
First generation leishmaniasis vaccines: A
review of field efficacy trials
Noazin,
Sassan et al.
Vaccine, 26(52),
6759 (
First generation candidate vaccines against leishmaniasis,
prepared using inactivated whole parasites as their main ingredient, were
considered as promising because of their relative ease of production and low
cost. These vaccines have been the subject of many investigations over several
decades and are the only leishmaniasis vaccine candidates which have undergone
phase 3 clinical trial evaluation. Although the studies demonstrated the safety
of the vaccines and several studies showed reasonable immunogenicity and some
indication of protection, an efficacious prophylactic vaccine is yet to be
identified. Despite this overall failure, these trials contributed
significantly to increasing knowledge on human leishmaniasis immunology. To
provide a collective view, this review discusses the methods and findings of
field efficacy trials of first generation leishmaniasis vaccine clinical trials
conducted in the Old and New Worlds.
One-step generation of double-allele gene
replacement mutants in Leishmania
donovani.
Ommen,
Gabi et al.
International Journal for Parasitology
Due to the apparent lack of sexual recombination in Leishmania
spp., gene replacement strategies in this diploid organism necessitate the
subsequent targeting of two gene alleles by using two constructs, bearing
different antibiotic resistance markers. This approach is time consuming and
often gives rise to spontaneous amplification of the targeted gene, nullifying
efforts to create functional null mutants. Here, authors show that by using two
homologous recombination constructs in a co-transfection of Leishmania donovani promastigotes, one
can obtain double-allele gene replacement mutants. This approach reduces the
time required for the generation of functional null mutants and the number of in vitro passage cycles, potentially
limiting culture-associated artefacts.
Bicyclic carbohydrates as antiprotozoal
bioactive for the treatment of infections caused by parasites
Sas , et al
Kemin
Pharma B.V.B.A. (
US Patent
7,125,854
The use of bicyclic carbohydrates for the treatment of
parasite infections is described. Different bicyclic carbohydrates have been
tested in vitro against a number of protozoa. These compounds also have been
screened against viruses, tumors, bacteria and fungi. Compound A1, a
thiophenyl-containing bicyclic carbohydrate possessed significant activity
against Trypanosoma brucei rhodiense, a parasite that causes the lethal
sleeping sickness. Compound A2 and Compound A3, bicyclic carbohydrates with
halogen containing aryl groups, possessed significant activity against
Leishmania donovani, a parasite that causes leishmaniasis. Bicyclic
carbohydrates in general, and Compound A1, Compound A2 and Compound A3 more
specifically, could be possible treatments for the sleeping sickness and
leishmaniasis in the future.
Suicidal mutant Leishmania vaccine
Chang , et al
US Patent
7,238,347
The invention discloses the use of a mutant Leishmania as a
suicidal vaccine wherein the mutant Leishmania is responsive to external
signals to become porphyric and commit suicidal cytolysis. The mutant can be
selected from natural Leishmania species or constructed by genetic engineering.
Practical serological assay for the
clinical diagnosis of leishmaniasis
Ryan;
Jeffrey R. et al.
The
US Patent
7,008,774
Methods for the diagnosis of visceral, cutaneous and canine
leishmaniasis in a subject suspected of being infected with the parasitic
protozoa Leishmania is disclosed. Disclosed are antibody-capture enzyme-linked
immunosorbent assays (ELISAs) for the detection of antibodies to Leishmania
parasite soluble antigens and antigen-capture ELISAs for the detection of
Leishmania parasite soluble antigens in host samples. Also disclosed are
immunodiagnostic kits for the detection of Leishmania parasite circulating
antigens or IgM and IgG antibodies in a sample from subject having visceral,
cutaneous or canine leishmaniasis. In these methods and kits, detection may be
done photometrically or visually. The methods and kits also allow the
visualization of Leishmania amastigotes or promastigotes in a sample.
Process for the in vitro culture of different stages of tissular parasites.
Lemesre;
Jean-Loup
Institut
de Recherche pour de Developpement, IRD, Paris,
US Patent
7,282,210
The invention provides a method for carrying out in vitro the complete developmental
sequence culture of tissular parasites, which includes culturing the parasites
in a totally defined culture medium which is an axenic . . . monophasic liquid
culture medium, free of serum and free of serum-derived or cell-derived macromolecules,
proteins and peptides. For obtaining amastigote forms, this medium is buffered
at a pH of 5.5 to 6.5 and has an osmolarity of at least 400 milliosmoles/kg of
liquid. For obtaining promastigote forms, the medium is buffered at a pH of 7
to 7.5 and has an osmolarity of at least 300 milliosmoles/kg liquid.
Application to the in vitro culture of different stages of tissular parasites
such as Leishmania, T. cruzi, and
hamatoprotozoa is also provided.
Biophosphonate compounds and methods for
bone resorption diseases, cancer, bone pain, immune disorders, and infectious
diseases.
Sanders;
John M. et al.
The Board
of Trustees of the
US Patent 7,358,361
Bisphosphonate compounds and related methods of making and
using are disclosed, including pyridinium-1-yl, quinolinium-1-yl, and related
compounds. The activity of compounds is disclosed in the context of functional
assays such as Leishmania major farnesyl
diphosphate synthase (FPPS) inhibition, Dictyostelium
discoideum growth inhibition, human gamma delta T cell activation, and bone
resorption. The applicability of bisphosphonate compounds in the context of
parasitic infections, is disclosed. Further potential applications of the
invention are disclosed regarding the treatment of one or more conditions such
as bone resorption disorders, cancer, bone pain, infectious diseases, and in
immunotherapy.
Cationic substituted benzofurans as
antimicrobial agents
Tidwell;
Richard R. et al
The
US Patent
7,417,158
A method of treating a Mycobacterium tuberculosis infection in
a subject in need thereof by administering to the subject an effective amount
of a cationic substituted benzofuran compound. Methods of treating microbial
infections, including infections from protozoan pathogens, such as Leishmania donovani, Trypanosoma brucei rhodesiense, a Trypanosoma cruzi, and Plasmodium falciparum, and fungal
pathogens, such as Candida albicans, Aspergillus fumigatus, and Cryptococcus neoformans, in a subject in
need thereof by administering to the subject an effective amount of a cationic
substituted benzofuran compound. Methods of synthesizing novel cationic
substituted benzofuran compounds and the novel compounds themselves.
Practical serological assay for the
clinical diagnosis of leishmaniasis.
Martin;
Samuel K et al.
The
Methods for the diagnosis of visceral, cutaneous and canine
leishmaniasis in a subject suspected of being infected with the parasitic
protozoa Leishmania is disclosed. Disclosed are antibody-capture enzyme-linked
immunosorbent assays (ELISAs) for the detection of antibodies to Leishmania
parasite soluble antigens and antigen-capture ELISAs for the detection of
Leishmania parasite soluble antigens in host samples. Also disclosed are
immunodiagnostic kits for the detection of Leishmania parasite circulating
antigens or IgM and IgG antibodies in a sample from subject having visceral,
cutaneous or canine leishmaniasis. In these methods and kits, detection may be
done photometrically or visually. The methods and kits also allow the
visualization of Leishmania amastigotes or promastigotes in a sample.
Therapeutic compositions and methods
useful in modulating protein tyrosine phosphatases.
Yi Taolin et al.
The
US7416723, 2008-08-26, 2002-09-09
A therapeutic composition containing a
pentavalent antimonial is provided. The pentavalent antimonial can be sodium
stibogluconate, levamisole, ketoconazole, and pentamidine and biological
equivalents of said compounds. Additionally, pentavalent antimonials that can
be used in accordance with the present invention may be any such compounds
which are anti-leishmaniasis agents. The therapeutic composition of this
embodiment contains an effective amount of pentavalent antimonial that can be
used in treating infectious diseases. The types of diseases that can be treated
with the present invention include, but are not limited to, the following:
diseases associated with PTPase activity, immune deficiency, cancer, infections
(such as viral infections), hepatitis B, and hepatitis C. The types of cancers
that the present embodiment can be used to treat include those such as
lymphoma, multiple myeloma, leukemia, melanoma, prostate cancer, breast cancer,
renal cancer, bladder cancer. The therapeutic composition enhances cytokine
activity. The therapeutic composition may include a cytokine, such as
interferon α, interferon β, interferon γ, or
granulocyte/macrophage colony stimulating factor.
Recombinant vectors based on the
modified vaccinia
Consejo
Superior De Investigaciones Científicas
WO07014977Al, 2007-02-08, 2006-07-28
The invention relates to recombinant
vectors based on the Modified Vaccinia Ankara (MVA) virus as vaccines against
leishmaniasis. The inventive vectors contain sequences encoding the LACK
protein, which are preferably inserted into the hemagglutinin locus of the
virus under the control of a promoter which enables the expression of same
throughout the infection cycle of the virus. The invention comprises stable,
safe vectors which elicit a strong immune response that provides protection
against leishmaniasis and which, as such, are particularly suitable for use in
vaccination against said disease, especially in humans, as well as in the
largest animal reservoir of said anthropozoonosis, i.e. dogs.
Anti-arthropod vector vaccines method
of selecting and uses thereof.
Valenzula
Jesus G and Belkaid,
US7388089, 2004-09-02
The invention provides methods of
selecting and uses of anti-arthropod vector vaccines to prevent Leishmaniasis.
The invention also provides compositions for vaccines to prevent Leishmaniasis.
Solid
pharmaceutical compositions containing miltefosine for oral administration in
the treatment of leishmaniasis.
Sauerbier D and
Engel J.
Asta
Medica AG,
CA2318260C,
2006-11-21, 1998-01-22
The invention relates to new solid pharmaceutical
compositions containing hexadecylphosphocholine (miltefosine) for oral
administration in the treatment of leishmaniasis, a process for the manufacture
of said pharmaceutical composition, a dosage scheme for oral administration of
said pharmaceutical composition in the treatment of leishmaniasis, and finally
a combination comprising said solid pharmaceutical composition, an
antiemeticum, and/or an antidiarrhoeal. A solid pharmaceutical composition for
the treatment of leishmaniasis in humans, wherein the pharmaceutical
composition comprises: hexadecylphos-phocholine (miltefosine); a
flow-controlling agent, or a lubricant, or both; and a filler; wherein the
pharmaceutical composition is formulated for oral administration to deliver a
total daily dosage in the range of about 10 to about 250 mg of
hexadecylphosphocholine over a period of time of about 2 to about 6 weeks; a nd
wherein said solid pharmaceutical composition is produced by a process that
does not use chloroform and is thereby free from the risk of contamination with
chloroform.
Phospholipid derivatives.
Engel J. and
Hilgard P.
Zentaris
GMBH,
CA2511753C, 2007-09-25, 1993-07-09
Compounds of the general formula wherein:
a quinuclidinio ring system linked via a carbon atom of the quinuclidinio ring
and in which the nitrogen atom optionally is substituted by a methyl group, or
a tropanio ring system linked via a carbon atom of the tropanio ring and in
which the nitrogen atom optionally is substituted by one or two methyl groups..
These phospholiped derivatives are useful for treating tumours, and protozoal
and fungal diseases. A pharmaceutical composition as defined in claim is for
the treatment of leishmaniasis.
Recombinant vectors based on the
modified vaccinia
Perez JE and
Larra AG
Consejo
Superior De
EP1916306Al, 2008-04-30, 2006-07-28
The invention relates to recombinant
vectors based on the Modified Vaccinia Ankara (MVA) virus as vaccines against
leishmaniasis. The inventive vectors contain sequences encoding the LACK
protein, which are preferably inserted into the hemagglutinin locus of the
virus under the control of a promoter, which enables the expression of same
throughout the infection cycle of the virus. The invention comprises stable,
safe vectors which elicit a strong immune response that provides protection
against leishmaniasis and which, as such, are particularly suitable for use in
vaccination against said disease, especially in humans, s well as in the
largest animal reservoir of said anthropozoonosis, i.e. dogs
The invention relates to recombinant
vectors based on the Modified Vaccinia Ankara (MVA) virus as vaccines against
leishmaniasis. The inventive vectors contain sequences encoding the LACK
protein, which are preferably inserted into the hemagglutinin locus of the
virus under the control of a promoter, which enables the expression of same
throughout the infection cycle of the virus. The invention comprises stable,
safe vectors which elicit a strong immune response that provides protection
against leishmaniasis and which, as such, are particularly suitable for use in
vaccination against said disease, especially in humans, s well as in the
largest animal reservoir of said anthropozoonosis, i.e. dogs.
Treating or reducing the risk of a
protozoal disease e.g. leishmaniasis, trypanosomiasis and malaria involves
administering at least one diindolylmethane-related indole optionally in
combination with at least one anti-protozoal agent.
Zeligs,
Michael A et al.
Bioresponse,
LLC,
WO/2008/057253, 15.05.2008, Application No. PCT/US2007/022649,
26.10.2007
The invention includes methods and compositions for the
treatment and prevention of protozoal parasitic infections utilizing
diindolylmethane-related indoles. Additive and synergistic interaction of
diindolylmethane-related indoles with other known anti-parasitic and
pro-apoptotic agents is believed to permit more effective therapy and prevention
of protozoal parasitic infections. The methods and compositions described
provide new treatment of protozoal parasitic diseases of mammals and birds
including malaria, leishmaniasis, trypanosomiasis, trichomoniasis, neosporosis
and coccidiosis.
Methods and materials for the detection of
leishmania infection.
Houghton,
Raymond L. et al.
Inbios
International Inc., Avenue South,
WO/2007/142695, 13.12.2007, Application
No. PCT/US2006/061699 06.12.2006
The invention provides rapid diagnostic assays for the
detection of Leishmania which can readily be used in the field, leading to more
rapid treatment. In certain embodiments, the inventive assays, including ELISA
and lateral flow assays, employ antibodies that may be effectively employed to
detect the Leishmania major antigen TSA, which is present in both promastigotes
grown in culture and in amastigotes. Such assays may be employed to detect the
presence of cutaneous leishmaniasis in a subject using scrapings, biopsies,
and/or aspirates taken from cutaneous lesions.
New recombinant vaccine for leishmaniasis
that allows the serologic distinction between vaccinated and infected canine
species with Leishmania, useful for enhancing immune response against
leishmaniasis.
Universidade
Federal de Minas Gerais
Universidade
Federal de Minas
WO/2008/009088, 24.01.2008, Application No. PCT/BR2007/000248,
20.07.2007
The present invention refers to the recombinant vaccine
against canine visceral leishmaniasis containing the recombinant A2 protein and
saponin, as an adjuvant, allowing the distinction between vaccinated and
infected animals through conventional ELISA or immunofluorescence tests that
employ antigens of promastigote forms of Lesihmania.
Leishmania as carriers
for the delivery of proteins and peptides.
Chang; Kwang-Poo et al.
US
Patent 7,261,887,
Methods for delivering potentially therapeutic or prophylactic
protein and peptide agents to mammalian cells are provided. The agents are
delivered by mutant trypanosomatid protozoa that have been genetically
manipulated to code for such protein or peptide agents. The mutant protozoa
additionally lack certain enzymes within the heme biosynthetic pathway, making
the mutants susceptible to porphyria and eventual lysis.
Method of combating infection.
John Burke
Philip et al.
Morvus
Technology Ltd.,
CN101282723 (A) 2008-10-08 WO2007026166 Application
number CN20068032152
20060904
A method of combating a parasitic protozoal infection of a
host organism, the method comprising administering tretazicar to the host
organism. Tretazicar is the compound 5-(aziridin-l-yl)-2,4-dinitrobenzamide (CB
1954), and the parasitic infection is preferably an infection of Trypanosoma cruzi, T. b. brucei,
Leishmania spp. particularly L. infantum,
Cryptosporidium or Giardia spp.
Genes associated with leishmania parasite virulence.
Yosser Ben
et al.
Inst
Pasteur
CN101274960 (A) 2008-10-01
The invention relates to the field of combating leishmaniases.
Said invention results from the isolation, from wild isolates of Leishmania major, of a protein-coding gene
known as LmPDI which has two regions that are identical to the sequence
(Cys-Gly-His-Cys) of the potential active site of the protein disulphide
isomerases (PDI). The LmPDI protein is predominantly expressed in the most
virulent isolates of the parasite. Said protein forms a novel therapeutic
target for developing anti-leishmaniasis medicaments and a novel element that
can be used in the composition of immunogenic, and possibly vaccinating,
preparations which are intended to protect a human or animal host against
Leishmania.
Nitroimidazole Compounds.
Jiricek
Jan et al.
Thomas
Hugo [SG]; Barry Clifton E [US]; Dowd Cynthia S [US
US2008275035 (A1) 2008-11-06 Application number:US20060097976
20061222
The invention relates to certain nitroimidazole compounds, which have interesting pharmaceutical properties. In particular, the compounds are useful in the treatment and/or prevention of infections such as those caused by Mycobacterium tuberculosis, Trypanosoma cruzi or Leishmania donovani. The invention also relates to pharmaceutical compositions containing the compounds, as well as processes for their preparation.
Papers published
on Leishmaniasis by the Central Drug Research Institute,
I.
Dube A, Singh N, Saxena A and Lakshmi V. - Antileishmanial
potential of a marine sponge, Haliclona exigua (Kirkpatrick) against
experimental visceral leishmaniasis. (2007) - Parasitology Research 101, 317-324
II.
Pandey Susmita, Suryawanshi SN, Nishi, Goyal Neena and Gupta
Suman Pandey Susmita, Suryawanshi SN, Nishi, Goyal Neena and Gupta Suman - Chemotherapy of
Leishmaniasis. Part V: Synthesis and in vitro bioevaluation of novel pyridinone
derivatives. (2007) - European Journal of Medicinal Chemistry 42, 669-674
III.
Singh N, Almeida R, Kothari H, Kumar P, Mandal G, Chatterjee M,
Venkatachalam S, Govind MK,
IV.
Suryawanshi SN, Bhatt BA, Pandey Susmita, Chandra Naveen and
Gupta Suman
- Chemotherapy of Leishmaniasis. Part VII: Synthesis and bioevaluation of
substituted terpenyl pyrimidines. (2007) - European Journal of Medicinal Chemistry
42, 1211-1217
V.
Suryawanshi SN, Pandey Susmita, Rashmirathi, Bhatt BA and Gupta
Suman -
Chemotherapy of Leishmaniasis Part VI: Synthesis and bioevaluation of some
novel terpenyl S,N- and N,N-acetals. (2007) - European Journal of Medicinal Chemistry
42, 511-516
VI.
Chandra, Naveen, Pandey Susmita, Ramesh, Suryawanshi SN &
Gupta Suman.
- Chemotherapy of leishmaniasis Part III: Synthesis and bioevaluation of novel
aryl substituted terpenyl pyrimidines as antileishmanial agents. (2006) - European J
Medicinal Chemistry :41, 779-85.
VII.
Garg, Ravendra and Dube, Anuradha - Animal models for vaccine studies for
visceral leishmaniasis (2006) - Indian Journal of Medical Research, 123, 439-454
VIII.
Garg, Ravendra; Gupta, Shraddha K; Tripathi, Parul; Hajela, K;
Sundar, S; Naik, S; Dube, Anuradha, - Leishmania donovani: identification of
stimulatory soluble antigenic proteins using cured human and hamster
lymphocytes for their prophylactic potential against visceral leishmaniasis (2006) - Vaccine, 24,
2900-2909.
IX.
Sharma Sharad Kumar, Dube, Anuradha, Ahmad Nadeem, Khan, Shazia,
Saleem, Iram, Garg, Ravendra and Mohammad, Owais - Non PC liposome entrapped promastigote
antigens elicit parasite specific CD8+ and CD4+ T-cell immune response and
protect hamsters against visceral leishmaniasis (2006) - Vaccine, 24,
1800-1810
X.
Garg, Ravendra , Srivastava, J.K , Pal, A. , Naik, S. &
Dube, Anuradha
- Isolation of integral membrane proteins of Leishmania promastigotes and
evaluation of their prophylactic potential in hamsters against experimental
visceral leishmaniasis, (2005) - Vaccine, Volume 23, Issue 9, 1189-1196.
XI.
Gupta, Suman , Ramesh , Sharma, S.C. & Srivastava, V.M.L. - Efficacy of picroliv
in combination with miltefosine, an orally effective antileishmanial drug
against experimental visceral leishmaniasis, (2005) - Acta Tropica, Volume 94, Issue 1,
41-47.
XII.
Pandey, Susmita , Suryawanshi, S.N. , Gupta, Suman &
Srivastava, V.M.L. - Chemotherapy of leishmaniasis part II: synthesis and bioevaluation
of substituted arylketene dithioacetals as antileishmanial agents, (2005) - European Journal of
Medicinal Chemistry, 40, 751-756
XIII.
Singh, Nasib , Mishra, Pushpesh Kumar , Kapil, Aruna , Arya,
Kamal Ram , Maurya, Rakesh & Dube, Anuradha - Efficacy of Desmodium gangeticum
extract and its fractions against experimental visceral leishmaniasis, (2005) - Journal of
Ethnopharmacology, Volume 98, Issues 1-2, 83-88.
XIV.
Gupta, Suman, Srivastava, V.M.L., Puri, Anju, Pandey, D., Haq,
W. - Adjunct
effect of immunostimulating hexapeptide analogous to human beta-casein fragment
(54-59) to sodium stibogluconate against experimental visceral leishmaniasis. (2004) - Immunopharmacology
And Immunotoxicology, Volume 26, Issue 3, August 2004, Pages 425-434.
XV.
Mishra, A., Dube, A. & Naik, S. - Immune responses in
normal Indian langur monkeys (Presbytis entellus) - A primate model for
visceral leishmaniasis. (2004) - Journal of Medical Primatology, Volume 33, Issue 2,
2004, Pages 65-69.
XVI.
Sharma, P., Rastogi, S., Bhatnagar, S., Srivastava, J.K., Dube,
A., Guru, P.Y., Kulshrestha, D.K., Mehrotra, B.N. & Dhawan, B.N. - Antileishmanial action
of Tephrosia purpurea linn, extract and its fractions against experimental
visceral leishmaniasis. (2003) - Drug Develop. Res., 60, 285-93.
XVII.
Gupta, S. & Tiwari, S. - Leishmania donovani infected hamster as
experimental model for visceral leishmaniasis. (2002) - J. Parasitic Dis.,
24, 211-13.
XVIII.
Tiwari, S. & Gupta, S. - Leishmaia donovani infected inbred golden
hamster as experimental model for visceral leishmaniasis. (2001) - J. Paraistic Dis.
XIX.
Ram. V.J. & Nath, M. - Progress in chemotherapy of leishmaniasis. (1996) - Curr. Med. Chem.,
3, 303-16.
XX.
Pal, A., Agarwal, A., Guru, P.Y. & Katiyar, J.C. - Enhanced efficacy of
liposomised pentamidine against experimental visceral leishmaniasis. (1995) - Journal of
Parasitic Diseases., 19, 45-48.
XXI.
Gupta, S., Srivastava, J.K., Pal, A., Katiyar, J.C., Saxena.
K.C., Dhawan, B.N. & Thakur, B.B. - Direct agglutination test and dot-ELISA in
the serodiagnosis of visceral leishmaniasis (kala-azar) a comparative study. (1994) - Serodiagnosis &
Immunotherapy Infec. Dis., 6, 154-58.
XXII.
Gupta, S., Srivastava, J.K., Pal, Ajai, Katiyar, J.C., Saxena,
K.C., Thakur, B.B. & Dhawan, B.N. - Direct agglutination test (DAT) and DOT-ELISA
in the serodiagnosis of visceral leishmaniasis (kala-azar). (1994) - Immunotherapy
Infect. Disease, 6, 154-58.
XXIII.
Anuradha, Pal, R., Zehra, K., Katiyar, J.C., Seth, N., Bhatia,
G. & Singh, R.K. - Presbytis entellus : A new non-human primate host for visceral
leishmaniasis. (1992) - Bull Wld. Hith. Organization,70, 63-72.
XXIV.
Chauhan, P.M.S. & Bhakuni, D.S. - Current Trends in the
Chemotherapy of Leishmaniasis. (1991) - Indian Drugs, 29, 1-11.
XXV.
Sen, A.B. & Guru, P.Y. - Chemotherapy of clinical & experimental
leishmaniasis. (1983) - Current status . Proceeding of
XXVI.
Singh N, Almeida R, Kothari H, Kumar P, Mandal G, Chatterjee M,
Venkatachalam S, Govind MK,
XXVII. Dube A., Singh N.,
Sundar S., Singh N. - Refractoriness to the treatment of sodium stibogluconate in
Indian kala-azar field isolates persist in in vitro and in vivo experimental
models (2005) - Parasitology
Research, 96, 216-223
XXVIII. Singh, N., Singh, R.T.
& Sundar, S. - Novel mechanism of drug resistance in kala-azar field isolates.
(2003) - J. Infect. Dis.,
188, 600-07.
XXIX.
Singh, N. - Is there true Sb (v) resistance in Indian Kala-azar field
isolates. (2002) - Curr. Sci., 83,
101-02.
XXX.
Gupta, N., Goyal, N., Bhatia, N., Kamboj, K.K. & Rastogi,
A.K. -
Identification of amastigote specific antigens of Leishmania donovani using
kalaazar patient sera. (2001) - Indian J. Expt. Biol., 39, 623-628.
XXXI.
Mukerji, K., Puri, A., Sahai, R., Saxena, R.P., Saxena, K.C.,
Dhawan, B.N. & Thakur, B.B. - Comparative potential of Leishmania donovani strains
MHOM/IN/80/Dd8 and MHOM/78/UR6 antigens in serodiagnosis of kala-azar by ELISA
and IFA. 24, 33-36. (2000) - J. Parasitic Dis.,24, 33-36.
XXXII. Gupta, S., Tiwari, S.,
Bagchi, A.K. & Katiyar, J.C. - A rising trend in kala-azar in
XXXIII. Singh, N., Sundar, S.,
Williams, F., Curran, M.D., Rastogi, A.K., Agrawal, S. & Middleton, D. - Molecular typing of
HLA class I and class II antigens in Indian kala-azar patients. (1997) - Trop. Med. Int.
Hlth., 2, 468-71.
XXXIV. Mukerji, K., Puri,A.,
Sahai, R., Saxena, R.P., Srivastava, J.K., Katiyar, J.C., Saxena, K.C., Dhawan,
B.N. & Thakur, B.B. - Evaluation of indirect fluorescent antibody (IFA) test for
Kala-azar for diagnostic potential in endemic areas. (1995) - Serodiag. &
Immunotherapy Infect. Dis., 8, 9-12.
XXXV. Gupta, S., Srivastava,
J.K., Pal, A., Katiyar, J.C., Saxena. K.C., Dhawan, B.N. & Thakur, B.B. - Direct agglutination
test and dot-ELISA in the serodiagnosis of visceral leishmaniasis (kala-azar) a
comparative study. (1994) - Serodiagnosis & Immunotherapy Infec. Dis., 6, 154-58.
XXXVI. Gupta, S., Srivastava,
J.K., Pal, Ajai, Katiyar, J.C., Saxena, K.C., Thakur, B.B. & Dhawan, B.N. - Direct agglutination
test (DAT) and DOT-ELISA in the serodiagnosis of visceral leishmaniasis
(kala-azar). (1994) - Immunotherapy Infect. Disease, 6, 154-58.
XXXVII. Gupta, S., Srivastava,
J.K. & Roy, S., Chandra, R., Srivastava, V.K. & Katiyar, J.C. - Evaluation of
enzyme-linkey immunosorbent assay in the diagerutic of kala-azar in Malda
district (West Bengal). (1993) - Indian J. Med. Res., 97, 242-45.
XXXVIII.
Chowdhury, Amita, Puri, Anju, Guru, P.Y., Saxena, R.P. &
Saxena, K.C.
- An indirect fluorescent antibody (IFA) test for the serodiagnosis of
kala-azar. (1992) - Journal of
Communicable Disease., 24(1), 32-36.
XXXIX. Katiyar, J.C., Anuradha
& Sharma, S. - Kala-azar : Current status a experimental chemotherapy. (1992) - Med. Res. Rev., 12,
473-504.
XL.
Katiyar, J.C., Anuradha & Sharma, S. - Kala-azar: Current
trends in chemotherapy. (1991) - Review Life Sci., 11, 79-94.
XLI.
Choudhry, A., Guru, P.Y., Saxena, R.P., Tandon, A. & Saxena,
K.C. -
Enzyme-linked immunosorbent assay (ELISA) in the diagnosis of kala-azar
patients from Bhadohi (
XLII.
Dutta, G.D.P. & Dutta, G.P. - Bulk products and comparison of
promastigote and amastigote antigens of Leishmania donovani for serodiagnosis
and value of serum protein and immunoglobulin changes in diagnosis of kalazar
cases. (1985) - J. Com. Dis., 17,
77-82.
XLIII.
Dutta, G.D.P. & Dutta, G.P. - Serodiagnosis of human kala-azar cases
by indirect fluorsecent antibody test. (1984) - Indian J. Parasitol., 8, 177.
Based on the information available in the Annual Reports of C.D.R.I.
Occurrence of any error or omission may kindly be overlooked.
Compiled by:
Wamiq F. Rahman